
SARS-CoV-2 Evasion of the Interferon System: Can We Restore Its Effectiveness?
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
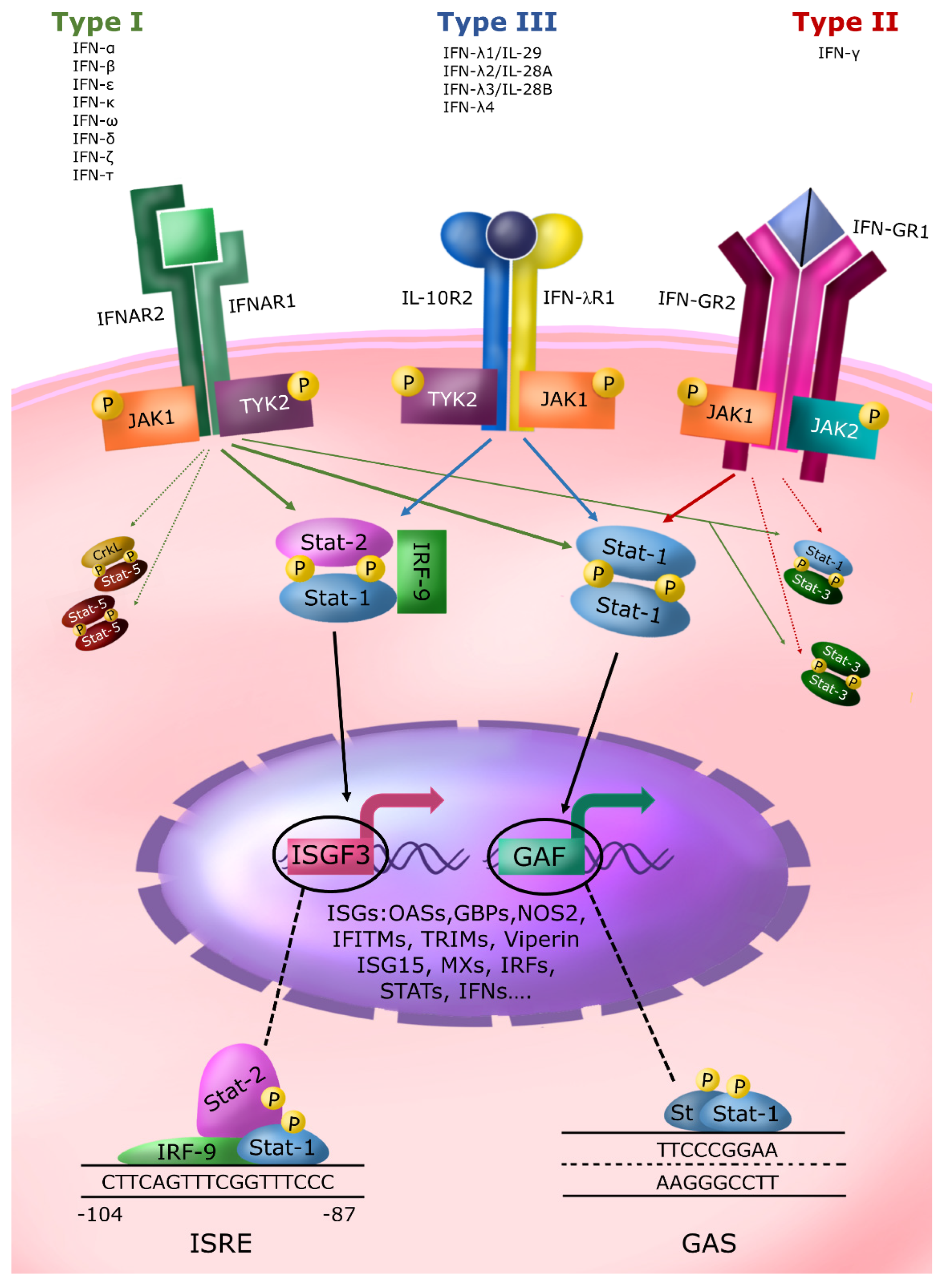
2. The IFN System and Its Relevance to Contrast Microbial Infections
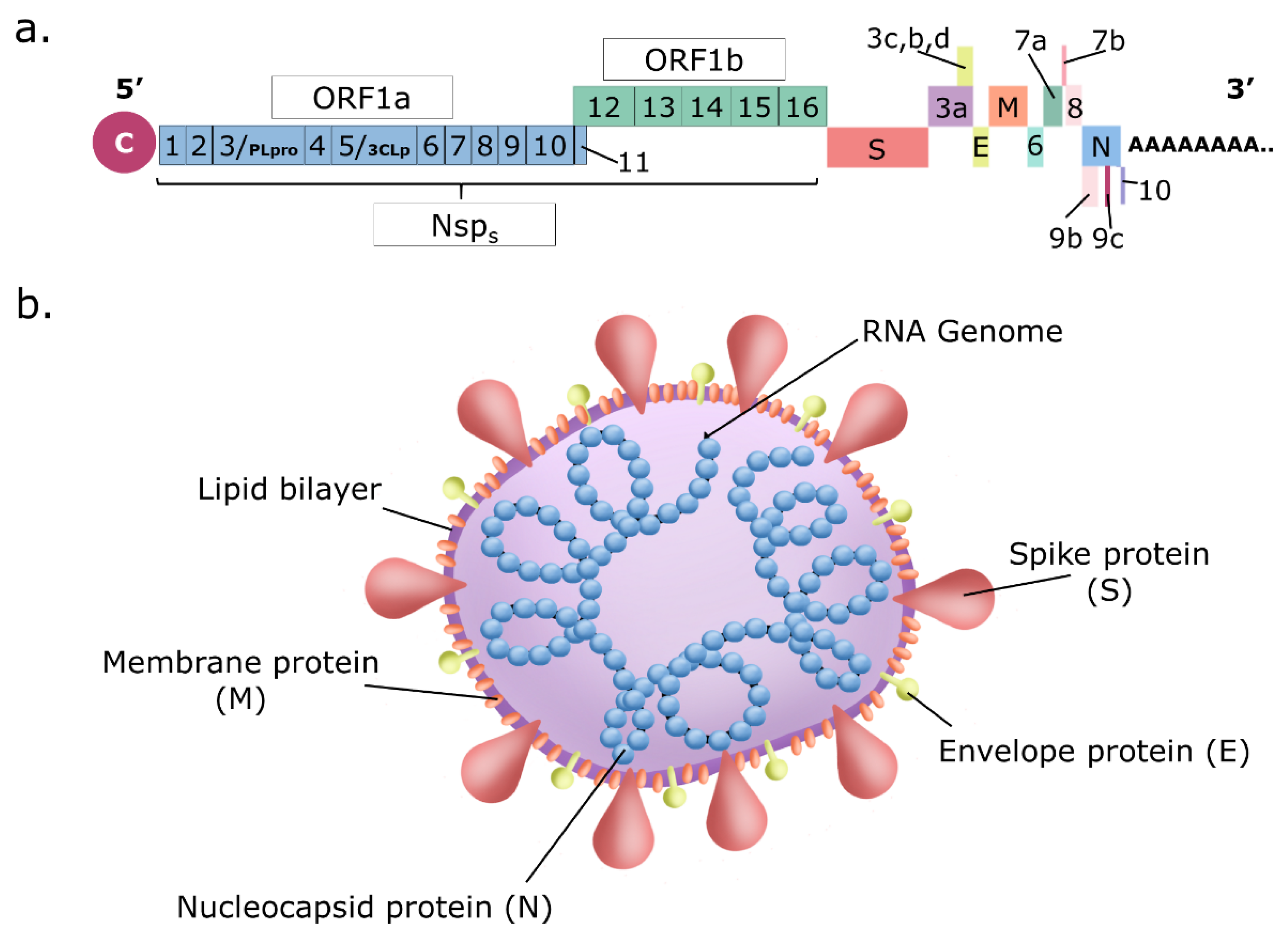
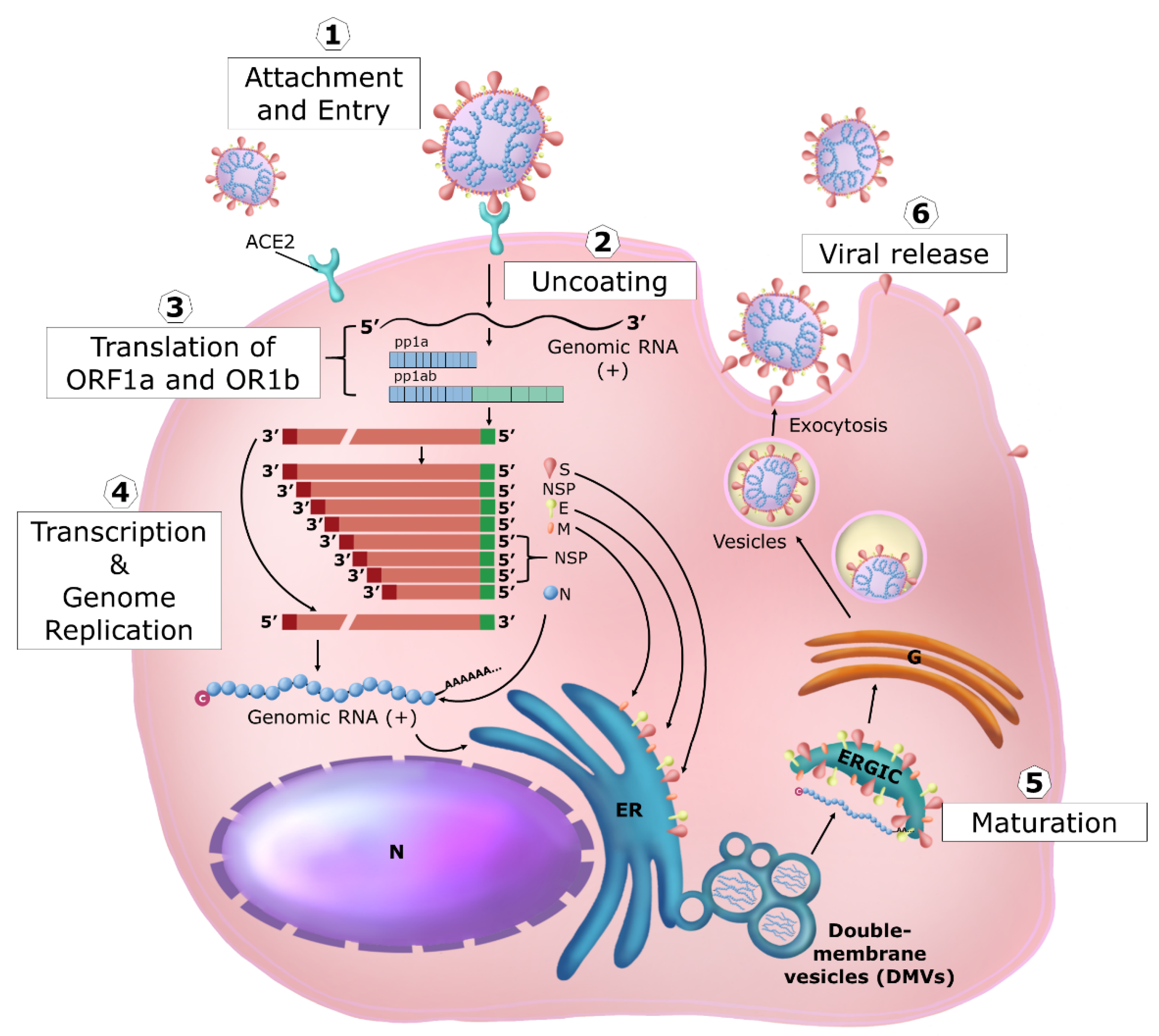
3. Coronavirus Replication
4. SARS-CoV-2-Induced Cytokine Storm
5. Genetic Background of the IFN System and SARS-CoV-2 Susceptibility
6. SARS-CoV-2 Evasion of the IFN System
7. Antiviral Treatments against COVID-19
8. Type I and III IFN Treatment Options
9. Can We Restore In Vivo IFNs Production and Susceptibility of Infected Cells?
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.; Wang, X.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.; Zhu, Y.; Li, B.; Huang, C.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P. Immune determinants of COVID-19 disease presentation and severity. Nat. Med. 2021, 27, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Alexandridi, M.; Muscolini, M.; Tassone, E.; Palermo, E.; Soultsioti, M.; Zevini, A. The global impact of the coronavirus pandemic. Cytokine Growth Factor Rev. 2020, 53, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leslie, M. A viral arsenal. Science 2022, 378, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J. Virol. 2020, 94, e00985-20. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.W.; Goeddel, D.V. Structure of the human immune interferon gene. Nature 1982, 298, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Lasfar, A.; Lewis-Antes, A.; Smirnov, S.V.; Anantha, S.; Abushahba, W.; Tian, B.; Reuhl, K.; Dickensheets, H.; Sheikh, F.; Donnelly, R.P.; et al. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res. 2006, 66, 4468–4477. [Google Scholar] [CrossRef]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Mamane, Y.; Heylbroeck, C.; Genin, P.; Algarte, M.; Servant, M.J.; LePage, C.; DeLuca, C.; Kwon, H.; Lin, R.; Hiscott, J. Interferon regulatory factors: The next generation. Gene 1999, 237, 1–14. [Google Scholar] [CrossRef]
- Solis, M.; Goubau, D.; Romieu-Mourez, R.; Genin, P.; Civas, A.; Hiscott, J. Distinct functions of IRF-3 and IRF-7 in IFN-alpha gene regulation and control of anti-tumor activity in primary macrophages. Biochem. Pharmacol. 2006, 72, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Osterlund, P.I.; Pietila, T.E.; Veckman, V.; Kotenko, S.V.; Julkunen, I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J. Immunol. 2007, 179, 3434–3442. [Google Scholar] [CrossRef] [PubMed]
- Panne, D.; Maniatis, T.; Harrison, S.C. An atomic model of the interferon-beta enhanceosome. Cell 2007, 129, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- De Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef]
- Casazza, R.L.; Lazear, H.M. Why Is IFN-lambda Less Inflammatory? One IRF Decides. Immunity 2019, 51, 415–417. [Google Scholar] [CrossRef]
- Schreiber, G. The molecular basis for differential type I interferon signaling. J. Biol. Chem. 2017, 292, 7285–7294. [Google Scholar] [CrossRef]
- Hemann, E.A.; Gale, M.J.; Savan, R. Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Darnell, J.E.J.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Kawasaki, B.T.; Howard, O.M.Z.; Gamero, A.M.; Farrar, W.L. Signaling by Cytokines. In The Cancer Handbook; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Levy, D.E.; Darnell, J.E.J. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Copeland, N.G.; Gilbert, D.J.; Schindler, C.; Zhong, Z.; Wen, Z.; Darnell, J.E.J.; Mui, A.L.; Miyajima, A.; Quelle, F.W.; Ihle, J.N. Distribution of the mammalian Stat gene family in mouse chromosomes. Genomics 1995, 29, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-lambda orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted disruption of the STAT-1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Meyts, I.; Casanova, J. Viral infections in humans and mice with genetic deficiencies of the type I IFN response pathway. Eur. J. Immunol. 2021, 51, 1039–1061. [Google Scholar] [CrossRef] [PubMed]
- Samarajiwa, S.A.; Forster, S.; Auchettl, K.; Hertzog, P.J. INTERFEROME: The database of interferon regulated genes. Nucleic Acids Res. 2009, 37, D852–D857. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, P.; Forster, S.; Samarajiwa, S. Systems biology of interferon responses. J. Interferon Cytokine Res. 2011, 31, 5–11. [Google Scholar] [CrossRef]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. Interferome v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013, 41, D1040–D1046. [Google Scholar] [CrossRef]
- Nakaya, T.; Sato, M.; Hata, N.; Asagiri, M.; Suemori, H.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem. Biophys. Res. Commun. 2001, 283, 1150–1156. [Google Scholar] [CrossRef]
- Tanaka, N.; Taniguchi, T. The interferon regulatory factors and oncogenesis. Semin. Cancer Biol. 2000, 10, 73–81. [Google Scholar] [CrossRef]
- Romeo, G.; Fiorucci, G.; Chiantore, M.V.; Percario, Z.A.; Vannucchi, S.; Affabris, E. IRF-1 as a negative regulator of cell proliferation. J. Interferon Cytokine Res. 2002, 22, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Rosain, J.; Neehus, A.; Manry, J.; Yang, R.; Le Pen, J.; Daher, W.; Liu, Z.; Chan, Y.; Tahuil, N.; Turel, O.; et al. Human IRF1 governs macrophagic IFN-gamma immunity to mycobacteria. Cell 2023, 186, 621–645.e33. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.; Chelbi-Alix, M.K. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 2011, 31, 145–158. [Google Scholar] [CrossRef] [PubMed]
- D’Cunha, J.; Knight, E.J.; Haas, A.L.; Truitt, R.L.; Borden, E.C. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc. Natl. Acad. Sci. USA 1996, 93, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Nakata, Y.; Ode, H.; Kubota, M.; Kasahara, T.; Matsuoka, K.; Sugimoto, A.; Imahashi, M.; Yokomaku, Y.; Iwatani, Y. Cellular APOBEC3A deaminase drives mutations in the SARS-CoV-2 genome. Nucleic Acids Res. 2023, 51, 783–795. [Google Scholar] [CrossRef]
- Fonteneau, J.; Larsson, M.; Beignon, A.; McKenna, K.; Dasilva, I.; Amara, A.; Liu, Y.; Lifson, J.D.; Littman, D.R.; Bhardwaj, N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 2004, 78, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Giannessi, F.; Percario, Z.A.; Affabris, E. The involvement of plasmacytoid cells in HIV infection and pathogenesis. Cytokine Growth Factor Rev. 2018, 40, 77–89. [Google Scholar] [CrossRef]
- Ronnblom, L.; Eloranta, M. The interferon signature in autoimmune diseases. Curr. Opin. Rheumatol. 2013, 25, 248–253. [Google Scholar] [CrossRef]
- Kretschmer, S.; Lee-Kirsch, M.A. Type I interferon-mediated autoinflammation and autoimmunity. Curr. Opin. Immunol. 2017, 49, 96–102. [Google Scholar] [CrossRef]
- Soper, A.; Kimura, I.; Nagaoka, S.; Konno, Y.; Yamamoto, K.; Koyanagi, Y.; Sato, K. Type I Interferon Responses by HIV-1 Infection: Association with Disease Progression and Control. Front. Immunol. 2018, 8, 1823. [Google Scholar] [CrossRef]
- Ng, C.S.; Kato, H.; Fujita, T. Fueling Type I Interferonopathies: Regulation and Function of Type I Interferon Antiviral Responses. J. Interferon Cytokine Res. 2019, 39, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.; Grivel, J.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: Role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and interferons: Who’s interfering with whom? Nat. Rev. Microbiol. 2015, 13, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Demoulins, T.; Abdallah, A.; Kettaf, N.; Baron, M.; Gerarduzzi, C.; Gauchat, D.; Gratton, S.; Sekaly, R. Reversible blockade of thymic output: An inherent part of TLR ligand-mediated immune response. J. Immunol. 2008, 181, 6757–6769. [Google Scholar] [CrossRef] [PubMed]
- Skelton, J.K.; Ortega-Prieto, A.M.; Kaye, S.; Jimenez-Guardeno, J.M.; Turner, J.; Malim, M.H.; Towers, G.J.; Dorner, M. Kinetics of Early Innate Immune Activation during HIV-1 Infection of Humanized Mice. J. Virol. 2019, 93, e02123-18. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Porritt, R.A.; Hertzog, P.J. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015, 36, 150–160. [Google Scholar] [CrossRef]
- Secombes, C.J.; Zou, J. Evolution of Interferons and Interferon Receptors. Front. Immunol. 2017, 8, 209. [Google Scholar] [CrossRef]
- Hertzog, P.J.; de Weerd, N.A. A structural “star” in interferon gamma signaling. Immunol. Cell Biol. 2019, 97, 442–444. [Google Scholar] [CrossRef]
- Savan, R. Post-transcriptional regulation of interferons and their signaling pathways. J. Interferon Cytokine Res. 2014, 34, 318–329. [Google Scholar] [CrossRef]
- Gale, M.J.; Savan, R. Introduction to the Special Issue. J. Interferon Cytokine Res. 2019, 39, 585. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 2249. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Norkin, L.C. Virology: Molecular Biology and Pathogenesis; ASM Press: Washington, DC, USA, 2010. [Google Scholar]
- Howley, P.M.; Knipe, D.M.; Whelan, S.P.J.; Freed, E.O. Fields Virology: RNA Viruses; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2022. [Google Scholar]
- Asea, A.A.; Kaur, P. Coronavirus Therapeutics–Volume I: Basic Science and Therapy Development; Springer: Cham, Switzerland, 2022. [Google Scholar]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef]
- Rehman, S.U.; Shafique, L.; Ihsan, A.; Liu, Q. Evolutionary Trajectory for the Emergence of Novel Coronavirus SARS-CoV-2. Pathogens 2020, 9, 240. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, F.; Chen, Y.; Plow, E.; Qin, J. Integrin mediates cell entry of the SARS-CoV-2 virus independent of cellular receptor ACE2. J. Biol. Chem. 2022, 298, 101710. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, A.; Filippini, F.; Bardelli, M.; Zani, A.; Chiodelli, P.; Messali, S.; Caruso, A.; Caccuri, F. SARS-CoV-2 Infects Human ACE2-Negative Endothelial Cells through an alpha(v)beta(3) Integrin-Mediated Endocytosis Even in the Presence of Vaccine-Elicited Neutralizing Antibodies. Viruses 2022, 14, 705. [Google Scholar] [CrossRef] [PubMed]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 2021, 372, 1306–1313. [Google Scholar] [CrossRef]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.; Leibundgut, M.; Thiel, V.; Muhlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Nelson, C.W.; Ardern, Z.; Goldberg, T.L.; Meng, C.; Kuo, C.; Ludwig, C.; Kolokotronis, S.; Wei, X. Dynamically evolving novel overlapping gene as a factor in the SARS-CoV-2 pandemic. eLife 2020, 9, e59633. [Google Scholar] [CrossRef]
- Chen, I.; Moriyama, M.; Chang, M.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Almendro-Vazquez, P.; Laguna-Goya, R.; Paz-Artal, E. Defending against SARS-CoV-2: The T cell perspective. Front. Immunol. 2023, 14, 1107803. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Ricci, G.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: Further advances in our understanding the role of specific chemokines involved. Cytokine Growth Factor Rev. 2021, 58, 82–91. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef]
- Cron, R.Q.; Goyal, G.; Chatham, W.W. Cytokine Storm Syndrome. Annu. Rev. Med. 2023, 74, 321–337. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.; Wang, Y.; Yuen, T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An ex Vivo Study with Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Qi, F.; Li, H.; Yang, Q.; Wang, H.; Wang, X.; Liu, X.; Zhao, J.; Liao, X.; Liu, Y.; et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Notari, S.; Gili, S.; Bordoni, V.; Casetti, R.; Cimini, E.; Tartaglia, E.; Mariotti, D.; Agrati, C.; Sacchi, A. Myeloid-Derived Suppressor Cells in COVID-19: The Paradox of Good. Front. Immunol. 2022, 13, 842949. [Google Scholar] [CrossRef] [PubMed]
- Ricci, D.; Etna, M.P.; Rizzo, F.; Sandini, S.; Severa, M.; Coccia, E.M. Innate Immune Response to SARS-CoV-2 Infection: From Cells to Soluble Mediators. Int. J. Mol. Sci. 2021, 22, 7017. [Google Scholar] [CrossRef] [PubMed]
- Combes, A.J.; Courau, T.; Kuhn, N.F.; Hu, K.H.; Ray, A.; Chen, W.S.; Chew, N.W.; Cleary, S.J.; Kushnoor, D.; Reeder, G.C.; et al. Global absence and targeting of protective immune states in severe COVID-19. Nature 2021, 591, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Severa, M.; Diotti, R.A.; Etna, M.P.; Rizzo, F.; Fiore, S.; Ricci, D.; Iannetta, M.; Sinigaglia, A.; Lodi, A.; Mancini, N.; et al. Differential plasmacytoid dendritic cell phenotype and type I Interferon response in asymptomatic and severe COVID-19 infection. PLoS Pathog. 2021, 17, e1009878. [Google Scholar] [CrossRef]
- Contoli, M.; Papi, A.; Tomassetti, L.; Rizzo, P.; Vieceli Dalla Sega, F.; Fortini, F.; Torsani, F.; Morandi, L.; Ronzoni, L.; Zucchetti, O.; et al. Blood Interferon-alpha Levels and Severity, Outcomes, and Inflammatory Profiles in Hospitalized COVID-19 Patients. Front. Immunol. 2021, 12, 648004. [Google Scholar] [CrossRef]
- Venet, M.; Ribeiro, M.S.; Decembre, E.; Bellomo, A.; Joshi, G.; Nuovo, C.; Villard, M.; Cluet, D.; Perret, M.; Pescamona, R.; et al. Severe COVID-19 patients have impaired plasmacytoid dendritic cell-mediated control of SARS-CoV-2. Nat. Commun. 2023, 14, 694. [Google Scholar] [CrossRef]
- Minkoff, J.M.; tenOever, B. Innate immune evasion strategies of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 178–194. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.; Su, H.C.; COVID Human Genetic Effort. A Global Effort to Define the Human Genetics of Protective Immunity to SARS-CoV-2 Infection. Cell 2020, 181, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 2020, 28, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Butler-Laporte, G.; Povysil, G.; Kosmicki, J.A.; Cirulli, E.T.; Drivas, T.; Furini, S.; Saad, C.; Schmidt, A.; Olszewski, P.; Korotko, U.; et al. Exome-wide association study to identify rare variants influencing COVID-19 outcomes: Results from the Host Genetics Initiative. PLoS Genet. 2022, 18, e1010367. [Google Scholar] [CrossRef]
- Lee, J.; Koepke, L.; Kirchhoff, F.; Sparrer, K.M.J. Interferon antagonists encoded by SARS-CoV-2 at a glance. Med. Microbiol. Immunol. 2023, 212, 125–131. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Guo, K.; Barrett, B.S.; Morrison, J.H.; Mickens, K.L.; Vladar, E.K.; Hasenkrug, K.J.; Poeschla, E.M.; Santiago, M.L. Interferon resistance of emerging SARS-CoV-2 variants. Proc. Natl. Acad. Sci. USA 2022, 119, e2203760119. [Google Scholar] [CrossRef]
- Domizio, J.D.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef]
- Chiale, C.; Greene, T.T.; Zuniga, E.I. Interferon induction, evasion, and paradoxical roles during SARS-CoV-2 infection. Immunol. Rev. 2022, 309, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Beyer, D.K.; Forero, A. Mechanisms of Antiviral Immune Evasion of SARS-CoV-2. J. Mol. Biol. 2022, 434, 167265. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.R.; Perlman, S. Immune dysregulation and immunopathology induced by SARS-CoV-2 and related coronaviruses—Are we our own worst enemy? Nat. Rev. Immunol. 2022, 22, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Quarleri, J.; Delpino, M.V. Type I and III IFN-mediated antiviral actions counteracted by SARS-CoV-2 proteins and host inherited factors. Cytokine Growth Factor Rev. 2021, 58, 55–65. [Google Scholar] [CrossRef]
- Palermo, E.; Di Carlo, D.; Sgarbanti, M.; Hiscott, J. Type I Interferons in COVID-19 Pathogenesis. Biology 2021, 10, 829. [Google Scholar] [CrossRef]
- Xu, D.; Biswal, M.; Neal, A.; Hai, R. Review Devil’s tools: SARS-CoV-2 antagonists against innate immunity. Curr. Res. Virol. Sci. 2021, 2, 100013. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-lambda Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890.e6. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical Role of Type III Interferon in Controlling SARS-CoV-2 Infection in Human Intestinal Epithelial Cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef]
- Andreakos, E.; Tsiodras, S. COVID-19: Lambda interferon against viral load and hyperinflammation. EMBO Mol. Med. 2020, 12, e12465. [Google Scholar] [CrossRef]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [PubMed]
- Iversen, M.B.; Ank, N.; Melchjorsen, J.; Paludan, S.R. Expression of type III interferon (IFN) in the vaginal mucosa is mediated primarily by dendritic cells and displays stronger dependence on NF-kappaB than type I IFNs. J. Virol. 2010, 84, 4579–4586. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Peng, L.; Park, J.J.; Hu, Y.; Devarkar, S.C.; Dong, M.B.; Shen, Q.; Wu, S.; Chen, S.; Lomakin, I.B.; et al. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Mol. Cell 2020, 80, 1055–1066.e6. [Google Scholar] [CrossRef]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef]
- Burke, J.M.; St Clair, L.A.; Perera, R.; Parker, R. SARS-CoV-2 infection triggers widespread host mRNA decay leading to an mRNA export block. RNA 2021, 27, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Swaim, C.D.; Canadeo, L.A.; Monte, K.J.; Khanna, S.; Lenschow, D.J.; Huibregtse, J.M. Modulation of Extracellular ISG15 Signaling by Pathogens and Viral Effector Proteins. Cell Rep. 2020, 31, 107772. [Google Scholar] [CrossRef]
- Loeb, K.R.; Haas, A.L. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J. Biol. Chem. 1992, 267, 7806–7813. [Google Scholar] [CrossRef]
- Liu, G.; Lee, J.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; van Gent, M.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef]
- Bogunovic, D.; Byun, M.; Durfee, L.A.; Abhyankar, A.; Sanal, O.; Mansouri, D.; Salem, S.; Radovanovic, I.; Grant, A.V.; Adimi, P.; et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science 2012, 337, 1684–1688. [Google Scholar] [CrossRef]
- Perng, Y.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Dzimianski, J.V.; Scholte, F.E.M.; Bergeron, E.; Pegan, S.D. ISG15: It’s Complicated. J. Mol. Biol. 2019, 431, 4203–4216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, J.; Yan, H.; Huang, J.; Wang, F.; Liu, T.; Zeng, L.; Zhou, F. ISGylation in Innate Antiviral Immunity and Pathogen Defense Responses: A Review. Front. Cell Dev. Biol. 2021, 9, 788410. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Reinert, J.T.; Pitha-Rowe, I.; Okumura, A.; Kellum, M.; Knobeloch, K.P.; Hassel, B.; Pitha, P.M. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell. Mol. Biol. 2006, 52, 29–41. [Google Scholar] [PubMed]
- Moustaqil, M.; Ollivier, E.; Chiu, H.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Cao, X. ISG15 secretion exacerbates inflammation in SARS-CoV-2 infection. Nat. Immunol. 2021, 22, 1360–1362. [Google Scholar] [CrossRef]
- Gold, I.M.; Reis, N.; Glaser, F.; Glickman, M.H. Coronaviral PLpro proteases and the immunomodulatory roles of conjugated versus free Interferon Stimulated Gene product-15 (ISG15). Semin. Cell Dev. Biol. 2022, 132, 16–26. [Google Scholar] [CrossRef]
- Mac Kain, A.; Maarifi, G.; Aicher, S.; Arhel, N.; Baidaliuk, A.; Munier, S.; Donati, F.; Vallet, T.; Tran, Q.D.; Hardy, A.; et al. Identification of DAXX as a restriction factor of SARS-CoV-2 through a CRISPR/Cas9 screen. Nat. Commun. 2022, 13, 2442. [Google Scholar] [CrossRef]
- Song, L.; Wang, D.; Abbas, G.; Li, M.; Cui, M.; Wang, J.; Lin, Z.; Zhang, X. The main protease of SARS-CoV-2 cleaves histone deacetylases and DCP1A, attenuating the immune defense of the interferon-stimulated genes. J. Biol. Chem. 2023, 299, 102990. [Google Scholar] [CrossRef]
- Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; Polishchuk, E.V.; Santoro, M.; Di Tullio, G.; Wilson, C.; Panariello, F.; Soares, V.C.; Dias, S.S.G.; et al. The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 2022, 606, 761–768. [Google Scholar] [CrossRef]
- Deng, J.; Zheng, Y.; Zheng, S.; Nan, M.; Han, L.; Zhang, J.; Jin, Y.; Pan, J.; Gao, C.; Wang, P. SARS-CoV-2 NSP7 inhibits type I and III IFN production by targeting the RIG-I/MDA5, TRIF, and STING signaling pathways. J. Med. Virol. 2023, 95, e28561. [Google Scholar] [CrossRef] [PubMed]
- Schindewolf, C.; Lokugamage, K.; Vu, M.N.; Johnson, B.A.; Scharton, D.; Plante, J.A.; Kalveram, B.; Crocquet-Valdes, P.A.; Sotcheff, S.; Jaworski, E.; et al. SARS-CoV-2 Uses Nonstructural Protein 16 To Evade Restriction by IFIT1 and IFIT3. J. Virol. 2023, 97, e0153222. [Google Scholar] [CrossRef] [PubMed]
- Sui, C.; Xiao, T.; Zhang, S.; Zeng, H.; Zheng, Y.; Liu, B.; Xu, G.; Gao, C.; Zhang, Z. SARS-CoV-2 NSP13 Inhibits Type I IFN Production by Degradation of TBK1 via p62-Dependent Selective Autophagy. J. Immunol. 2022, 208, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhuang, M.; Han, L.; Zhang, J.; Nan, M.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther 2020, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, S.; Zheng, Z.; Huang, Y.; Li, W.; Xu, Z.; Wang, Y. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef]
- Louis, C.; Burns, C.; Wicks, I. TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front. Immunol. 2018, 9, 434. [Google Scholar] [CrossRef]
- Cheng, N.; Liu, M.; Li, W.; Sun, B.; Liu, D.; Wang, G.; Shi, J.; Li, L. Protein post-translational modification in SARS-CoV-2 and host interaction. Front. Immunol. 2023, 13, 1068449. [Google Scholar] [CrossRef]
- Bhat, S.; Rishi, P.; Chadha, V.D. Understanding the epigenetic mechanisms in SARS-CoV-2 infection and potential therapeutic approaches. Virus Res. 2022, 318, 198853. [Google Scholar] [CrossRef]
- Su, J.; Shen, S.; Hu, Y.; Chen, S.; Cheng, L.; Cai, Y.; Wei, W.; Wang, Y.; Rui, Y.; Yu, X. SARS-CoV-2 ORF3a inhibits cGAS-STING-mediated autophagy flux and antiviral function. J. Med. Virol. 2023, 95, e28175. [Google Scholar] [CrossRef]
- Timilsina, U.; Umthong, S.; Ivey, E.B.; Waxman, B.; Stavrou, S. SARS-CoV-2 ORF7a potently inhibits the antiviral effect of the host factor SERINC5. Nat. Commun. 2022, 13, 2935. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Diehl, W.E.; Guney, M.H.; Vanzo, T.; Kyawe, P.P.; White, J.M.; Pizzato, M.; Luban, J. Influence of Different Glycoproteins and of the Virion Core on SERINC5 Antiviral Activity. Viruses 2021, 13, 1279. [Google Scholar] [CrossRef]
- Mangino, G.; Famiglietti, M.; Capone, C.; Veroni, C.; Percario, Z.A.; Leone, S.; Fiorucci, G.; Lulf, S.; Romeo, G.; Agresti, C.; et al. HIV-1 Myristoylated Nef Treatment of Murine Microglial Cells Activates Inducible Nitric Oxide Synthase, NO2 Production and Neurotoxic Activity. PLoS ONE 2015, 10, e0130189. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; USFQ-COVID19 Consortium; Nakagawa, S.; et al. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef]
- Han, L.; Zhuang, M.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.; Zhang, X.; Gao, C.; Wang, P. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, K.; Qin, B.; Olieric, V.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 Orf9b in complex with human TOM70 suggests unusual virus-host interactions. Nat. Commun. 2021, 12, 2843. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Chazal, N. Coronavirus, the King Who Wanted More Than a Crown: From Common to the Highly Pathogenic SARS-CoV-2, Is the Key in the Accessory Genes? Front. Microbiol. 2021, 12, 682603. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.T.; Cheung, V.; Salamango, D.J. Decoupling SARS-CoV-2 ORF6 localization and interferon antagonism. J. Cell Sci. 2022, 135, jcs259666. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.; Guedan, A.; Yap, M.W.; Young, G.R.; Harvey, R.; Stoye, J.P.; Bishop, K.N. SARS-CoV-2 ORF6 disrupts innate immune signalling by inhibiting cellular mRNA export. PLoS Pathog. 2022, 18, e1010349. [Google Scholar] [CrossRef] [PubMed]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Becker, M.E.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S.; et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 2021, 81, 2656–2668.e8. [Google Scholar] [CrossRef]
- Ali, M.; Rajurkar, J.; Majumder, P.; Jha, M.P.; Sarkar, R.; Mapa, K. Possible Therapeutic Intervention Strategies for COVID-19 by Manipulating the Cellular Proteostasis Network. Adv. Exp. Med. Biol. 2021, 1352, 125–147. [Google Scholar]
- Sarkar, K.; Sil, P.C. Potential Drug Strategies to Target Coronaviruses. Adv. Exp. Med. Biol. 2021, 1352, 111–124. [Google Scholar]
- Nie, Z.; Sun, T.; Zhao, F. Safety and Efficacy of Antiviral Drugs for the Treatment of COVID-19: A Systematic Review. Infect. Drug Resist. 2022, 15, 4457–4466. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Jochmans, D.; Vangeel, L.; De Jonghe, S.; Augustijns, P.; Mols, R.; Weynand, B.; Wattanakul, T.; Hoglund, R.M.; et al. The oral protease inhibitor (PF-07321332) protects Syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat. Commun. 2022, 13, 719. [Google Scholar] [CrossRef]
- Drozdzal, S.; Rosik, J.; Lechowicz, K.; Machaj, F.; Szostak, B.; Przybycinski, J.; Lorzadeh, S.; Kotfis, K.; Ghavami, S.; Los, M.J. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updates 2021, 59, 100794. [Google Scholar] [CrossRef] [PubMed]
- Najjar-Debbiny, R.; Gronich, N.; Weber, G.; Khoury, J.; Amar, M.; Stein, N.; Goldstein, L.H.; Saliba, W. Effectiveness of Paxlovid in Reducing Severe Coronavirus Disease 2019 and Mortality in High-Risk Patients. Clin. Infect. Dis. 2023, 76, e342–e349. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Ma, P.; Wang, M.; Cheng, Y.; Zhou, M.; Ye, L.; Feng, Z.; Zhang, C. Efficacy and safety of Paxlovid for COVID-19: A meta-analysis. J. Infect. 2023, 86, 66–117. [Google Scholar] [CrossRef] [PubMed]
- Bojkova, D.; Stack, R.; Rothenburger, T.; Kandler, J.D.; Ciesek, S.; Wass, M.N.; Michaelis, M.; Cinatl, J., Jr. Synergism of interferon-beta with antiviral drugs against SARS-CoV-2 variants. J. Infect. 2022, 85, 573–607. [Google Scholar] [CrossRef]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.; et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 2023, 613, 558–564. [Google Scholar] [CrossRef]
- Pardo, J.; Shukla, A.M.; Chamarthi, G.; Gupte, A. The journey of remdesivir: From Ebola to COVID-19. Drugs Context 2020, 9, 2020-4-14. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Cao, Z.; Gao, W.; Bao, H.; Feng, H.; Mei, S.; Chen, P.; Gao, Y.; Cui, Z.; Zhang, Q.; Meng, X.; et al. VV116 versus Nirmatrelvir-Ritonavir for Oral Treatment of COVID-19. N. Engl. J. Med. 2023, 388, 406–417. [Google Scholar] [CrossRef]
- Tam, A.R.; Zhang, R.R.; Lung, K.; Liu, R.; Leung, K.; Liu, D.; Fan, Y.; Lu, L.; Lam, A.H.; Chung, T.W.; et al. Early Treatment of High-Risk Hospitalized Coronavirus Disease 2019 (COVID-19) Patients with a Combination of Interferon Beta-1b and Remdesivir: A Phase 2 Open-label Randomized Controlled Trial. Clin. Infect. Dis. 2023, 76, e216–e226. [Google Scholar] [CrossRef]
- Galani, I.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, V.; Shannon, C.P.; Wei, X.; Xiang, X.; Wang, X.; Wang, Z.; Tebbutt, S.J.; Kollmann, T.R.; Fish, E.N. Corrigendum: Interferon-alpha2b Treatment for COVID-19. Front. Immunol. 2020, 11, 615275. [Google Scholar] [CrossRef]
- Monk, P.D.; Marsden, R.J.; Tear, V.J.; Brookes, J.; Batten, T.N.; Mankowski, M.; Gabbay, F.J.; Davies, D.E.; Holgate, S.T.; Ho, L.; et al. Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2021, 9, 196–206. [Google Scholar] [CrossRef]
- Schreiber, G. The Role of Type I Interferons in the Pathogenesis and Treatment of COVID-19. Front. Immunol. 2020, 11, 595739. [Google Scholar] [CrossRef]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef]
- Lee, J.S.; Shin, E. The type I interferon response in COVID-19: Implications for treatment. Nat. Rev. Immunol. 2020, 20, 585–586. [Google Scholar] [CrossRef]
- Lu, L.; Feng, P.; Yu, M.; Chen, M.; Lin, A.J.; Chen, J.L.; Yu, L.H. Current utilization of interferon alpha for the treatment of coronavirus disease 2019: A comprehensive review. Cytokine Growth Factor Rev. 2022, 63, 34–43. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.J.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Forero, A.; Ozarkar, S.; Li, H.; Lee, C.H.; Hemann, E.A.; Nadjsombati, M.S.; Hendricks, M.R.; So, L.; Green, R.; Roy, C.N.; et al. Differential Activation of the Transcription Factor IRF1 Underlies the Distinct Immune Responses Elicited by Type I and Type III Interferons. Immunity 2019, 51, 451–464.e6. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Hartmann, R.; Wack, A. IFNlambda is a potent anti-influenza therapeutic without the inflammatory side effects of IFNalpha treatment. EMBO Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Kandel, C.; Biondi, M.J.; Kozak, R.A.; Zahoor, M.A.; Lemieux, C.; Borgia, S.M.; Boggild, A.K.; Powis, J.; McCready, J.; et al. Peginterferon lambda for the treatment of outpatients with COVID-19: A phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021, 9, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Santer, D.M.; Li, D.; Ghosheh, Y.; Zahoor, M.A.; Prajapati, D.; Hansen, B.E.; Tyrrell, D.L.J.; Feld, J.J.; Gehring, A.J. Interferon-lambda treatment accelerates SARS-CoV-2 clearance despite age-related delays in the induction of T cell immunity. Nat. Commun. 2022, 13, 6992. [Google Scholar] [CrossRef] [PubMed]
- Reis, G.; Moreira Silva, E.A.S.; Medeiros Silva, D.C.; Thabane, L.; Campos, V.H.S.; Ferreira, T.S.; Santos, C.V.Q.; Nogueira, A.M.R.; Almeida, A.P.F.G.; Savassi, L.C.M.; et al. Early Treatment with Pegylated Interferon Lambda for COVID-19. N. Engl. J. Med. 2023, 388, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- La Rosee, F.; Bremer, H.C.; Gehrke, I.; Kehr, A.; Hochhaus, A.; Birndt, S.; Fellhauer, M.; Henkes, M.; Kumle, B.; Russo, S.G.; et al. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia 2020, 34, 1805–1815. [Google Scholar] [CrossRef]
- Neubauer, A.; Wiesmann, T.; Vogelmeier, C.F.; Mack, E.; Skevaki, C.; Gaik, C.; Keller, C.; Figiel, J.; Sohlbach, K.; Rolfes, C.; et al. Ruxolitinib for the treatment of SARS-CoV-2 induced acute respiratory distress syndrome (ARDS). Leukemia 2020, 34, 2276–2278. [Google Scholar] [CrossRef]
- Bode, J.G.; Brenndorfer, E.D.; Haussinger, D. Subversion of innate host antiviral strategies by the hepatitis C virus. Arch. Biochem. Biophys. 2007, 462, 254–265. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sacchi, A.; Giannessi, F.; Sabatini, A.; Percario, Z.A.; Affabris, E. SARS-CoV-2 Evasion of the Interferon System: Can We Restore Its Effectiveness? Int. J. Mol. Sci. 2023, 24, 9353. https://doi.org/10.3390/ijms24119353
Sacchi A, Giannessi F, Sabatini A, Percario ZA, Affabris E. SARS-CoV-2 Evasion of the Interferon System: Can We Restore Its Effectiveness? International Journal of Molecular Sciences. 2023; 24(11):9353. https://doi.org/10.3390/ijms24119353
Chicago/Turabian StyleSacchi, Alessandra, Flavia Giannessi, Andrea Sabatini, Zulema Antonia Percario, and Elisabetta Affabris. 2023. "SARS-CoV-2 Evasion of the Interferon System: Can We Restore Its Effectiveness?" International Journal of Molecular Sciences 24, no. 11: 9353. https://doi.org/10.3390/ijms24119353