Article Text

Statistics from Altmetric.com
- arrhythmogenic right ventricular dysplasia
- cardiac magnetic resonance (CMR) imaging
- educational goals and objectives
- ventricular fibrillation
Learning objectives
To review the genetic background and the phenotypic features including clinical, ECG, arrhythmic and imaging findings of arrhythmogenic left ventricular cardiomyopathy (ALVC).
To address the 2020 international criteria for diagnosis of arrhythmogenic cardiomyopathy (the ‘Padua criteria’), including the diagnostic criteria for ALVC and differential diagnosis with major phenocopies.
To evaluate the current risk stratification and therapy for ALVC, with particular reference to indications for implantation of a defibrillator.
Introduction
Arrhythmogenic cardiomyopathy (ACM) is a genetic heart muscle disease characterised by substitution of the ventricular myocardium by fibrofatty tissue.1 The disease was originally termed ‘arrhythmogenic right ventricular (dysplasia/) cardiomyopathy’ (ARVC) to define a condition which distinctively affected the right ventricle (RV) and predisposed to potentially fatal ventricular arrhythmias, particularly in young individuals and athletes.2–4 New insights arising from postmortem investigations, genotype–phenotype correlation studies and myocardial tissue characterisation by contrast-enhanced cardiac magnetic resonance (CMR) led to increased awareness that the disease often also involves the left ventricle (LV).5–11 The current designation of ‘arrhythmogenic cardiomyopathy’ better reflects the evolving concept of a heart muscle disease affecting both ventricles, with some phenotypic variants characterised by a parallel or predominant involvement of the LV. The adjective ‘arrhythmogenic’ refers to the distinctive clinical propensity of the disease to cause ventricular arrhythmias in relation to the fibrofatty scar tissue, which is a recognised arrhythmogenic myocardial substrate. Patients with a long-standing disease may develop RV or biventricular dysfunction.4
This definition of ACM substantially differs from that provided by the 2019 expert consensus statement endorsed by the Heart Rhythm Society (HRS),12 that is, a non-specific entity which encompasses a heterogeneous group of heart muscle disorders, including ‘systemic’ diseases such as sarcoidosis and amyloidosis; ‘inflammatory’ diseases such as myocarditis; ‘infectious’ diseases such as Chagas disease; and ‘genetic’ diseases such as desmosomal and non-desmosomal forms; and ‘ion channel diseases’. According to the HRS document, the vague common denominator of this miscellaneous group of ‘arrhythmogenic cardiomyopathies’ was the ‘clinical presentation with symptoms or documentation of atrial fibrillation, conduction disease, and/or RV and/or LV arrhythmia’. The HRS definition appears inappropriate because ACM is a nosographically distinct condition characterised by typical cardiomyopathic features.
The current classification of ACM includes the following major phenotypes: (1) the original ARVC phenotype with predominant RV involvement and no LV abnormalities (‘dominant-right’ variant); (2) the phenotypic variant characterised by the equal involvement of both ventricles (‘biventricular’); and (3) the phenotypic variant characterised by predominant LV involvement with no or minor RV abnormalities (‘dominant-left’), also referred to as ‘arrhythmogenic left ventricular cardiomyopathy (ALVC)’.13 14
In 2000 we had the privilege to write a commissioned article for Education in Heart on ‘Arrhythmogenic right ventricular cardiomyopathy: pathogenesis, diagnosis and therapy’.15 Twenty years later, we are proud to provide for the same educational section of Heart with an updated article on ‘arrhythmogenic left ventricular cardiomyopathy (ALVC)’, which represents the more recently characterised variant at the other end of the broad spectrum of the phenotypic expression of ACM.
The present article addresses the available knowledge on the genetic background, phenotypic features including clinical, ECG, arrhythmic and imaging findings, risk stratification and therapy of ALVC. The 2020 international diagnostic criteria for ACM (the ‘Padua criteria’), which included criteria for diagnosis of left-sided phenotypic variants, will be also discussed.14
Phenotype
The increasing availability of pathological and clinical studies on patients with left-sided phenotypic variants of ACM has provided significant insights into the prevalence and the clinical manifestations of ALVC.5–23
In the seminal pathological study on the heart of sudden cardiac death (SCD) victims (or patients undergoing heart transplantation) with ACM, evidence of macroscopic and/or histological LV involvement was found in 76% of cases, a figure showing that the disease affects the LV in the majority of cases.6 LV lesions usually affected both the LV free wall and the septum, more often regionally, with a predilection for the posteroseptal and posterolateral areas. The LV lesion was most often located in the outer layer of the wall musculature and consisted of large areas of subepicardial (and less frequently mid-myocardial) fibrofatty myocardium replacement, similarly to the RV lesions. A more recent autopsy study confirmed that LV involvement was observed in majority (87%) of ACM-related SCD victims (20% athletes) and appeared as an isolated pathological finding (ALVC) in almost a fifth of cases.10 The most common areas of fibrofatty infiltration were the LV posterobasal (68%) and anterolateral (58%) walls. Postmortem genetic testing yielded pathogenic variants in ACM-related genes in 25% decedents.
CMR studies in living patients fulfilling the 2010 International Task Force (ITF) criteria have consistently shown that LV involvement in terms of morphofunctional (LV global or regional systolic dysfunction) and/or structural (LV late gadolinium enhancement (LGE)) abnormalities is identified in more than half of patients.9 22 24
 According to the available findings of clinical studies, phenotypic features of left-sided ACM include the following (figure 1): (1) ECG abnormalities such as low-amplitude QRS complexes (peak to peak <0.5 mV) in limb leads and T-wave inversion or flattening in the lateral (or inferolateral) leads, although the ECG is often normal; (2) ventricular arrhythmias with a right bundle branch block (RBBB) morphology of the ectopic QRS (denoting the origin from the LV); (3) normal or slightly depressed LV systolic function with no (or mild) dilatation; (4) large amount of myocardial fibrosis evidenced by contrast-enhanced CMR as LGE; and (5) ‘non-ischemic’ pattern of LGE, predominantly involving the subepicardial layers of the inferior and the inferolateral regions. This last finding is in agreement with the notion that the wave-front of myocardial loss and fibrofatty replacement in the LV wall proceeds from the epicardium to the endocardium, with scar tissue mostly confined to the epicardial layers of the wall. The subendocardial layer which mostly accounts for the regional LV wall contractility is usually conserved, a factor which explains the normal results of echocardiography, cine-CMR and left ventriculography in a sizeable proportion of patients, due to the preservation of global LV systolic function and the lack of wall motion abnormalities.22 25 As a corollary, an imaging approach limited to a mere evaluation of the LV function, either global or
According to the available findings of clinical studies, phenotypic features of left-sided ACM include the following (figure 1): (1) ECG abnormalities such as low-amplitude QRS complexes (peak to peak <0.5 mV) in limb leads and T-wave inversion or flattening in the lateral (or inferolateral) leads, although the ECG is often normal; (2) ventricular arrhythmias with a right bundle branch block (RBBB) morphology of the ectopic QRS (denoting the origin from the LV); (3) normal or slightly depressed LV systolic function with no (or mild) dilatation; (4) large amount of myocardial fibrosis evidenced by contrast-enhanced CMR as LGE; and (5) ‘non-ischemic’ pattern of LGE, predominantly involving the subepicardial layers of the inferior and the inferolateral regions. This last finding is in agreement with the notion that the wave-front of myocardial loss and fibrofatty replacement in the LV wall proceeds from the epicardium to the endocardium, with scar tissue mostly confined to the epicardial layers of the wall. The subendocardial layer which mostly accounts for the regional LV wall contractility is usually conserved, a factor which explains the normal results of echocardiography, cine-CMR and left ventriculography in a sizeable proportion of patients, due to the preservation of global LV systolic function and the lack of wall motion abnormalities.22 25 As a corollary, an imaging approach limited to a mere evaluation of the LV function, either global or  regional, by echocardiography, angiography or cine-CMR appears insufficient to detect LV involvement and characterise the LV phenotype in patients with ACM.
regional, by echocardiography, angiography or cine-CMR appears insufficient to detect LV involvement and characterise the LV phenotype in patients with ACM.

Phenotypic features of arrhythmogenic left ventricular cardiomyopathy. ECG strips showing low QRS voltages (<0.5 mV) in the limb leads (arrow) (A), negative T-waves in leads V4–V6 (arrows) (B), premature ventricular beat with a right bundle branch block and superior axis (C), and ventricular tachycardia with a right bundle branch block and inferior axis morphology (D). Two-dimensional echocardiogram (diastolic frame in four-chamber view) showing a non-dilated and normokinetic (not shown) left ventricle (E). Cardiac magnetic resonance (diastolic frame of four-chamber view on postcontrast sequences) showing diffuse late gadolinium enhancement involving the subepicardial layer of the left ventricle free wall and the septum (F). Histological panoramic view of the posterolateral left ventricular wall showing replacement-type myocardial fibrosis mostly involving the subepicardial layer (trichrome stain) (G).
 Genotype
Genotype

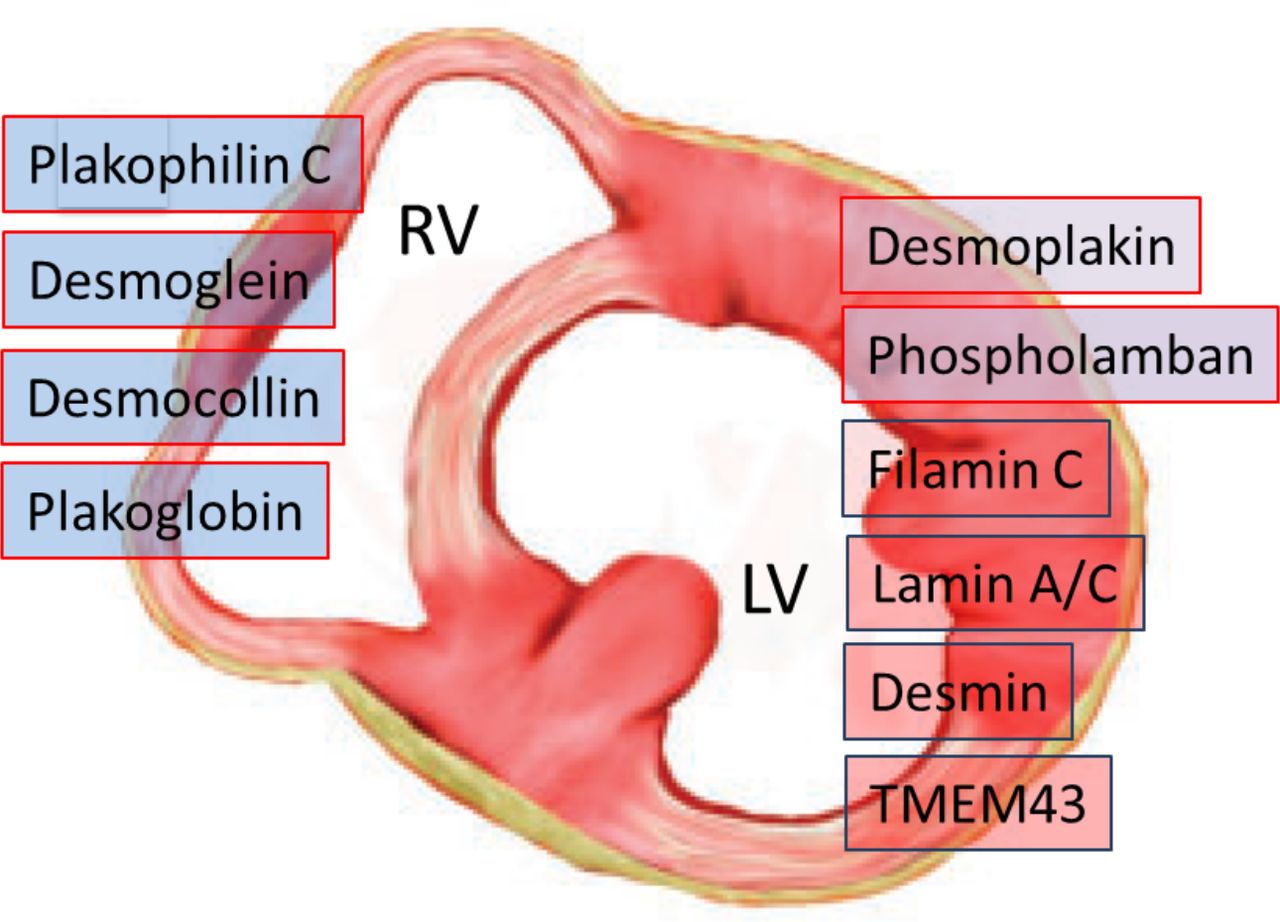
Most of the pathogenic mutations of ACM affect genes encoding structural proteins that are involved in the organisation of intercellular junctions.1 4 26 27 These include genes encoding cardiac desmosomal proteins most frequently, such as plakophilin (PKP2), desmoplakin (DSP), desmoglein (DSG2) and desmocollin (DSC2), and rarely (<1%) genes encoding for adherens junctional proteins of the ‘area composita’, such as α-T-catenin (CTNNA3) and N-cadherin (CDH2). ACM-causing mutations have also been found in non-desmosomal genes such as phospholamban (PLN), filamin C (FLNC), desmin (DES), titin (TTN) and lamin A/C (LMNA), which are associated with other cardiomyopathies, such as dilated cardiomyopathy (DCM) and neuromuscular cardiomyopathies, and may lead to overlapping phenotypes. Causal mutations in non-desmosomal genes, such as transmembrane protein 43 (TMEM43) and transforming growth factor beta-3 (TGFß-3) genes, have been uncommonly identified. Genotype–phenotype correlation studies have demonstrated that mutations in DSP, PLN and FLNC genes are the most common gene defects causing left-sided ACM (figure 2).1 11 17–23

Disease-causing genetic defects in arrhythmogenic cardiomyopathy according to predominant ventricular involvement. Mutations in DSP, PLN and FLNC genes are the most common gene defects causing arrhythmogenic left ventricular cardiomyopathy. LV, left ventricle; RV, right ventricle.
Desmoplakin is one of the major components of the desmosome that connects the cytoskeletal networks to the cellular membrane, specifically linking the desmocollins and desmogleins to the intermediate filament through interaction with plakoglobin and plakophilin. A number of human DSP gene mutations have been linked with ACM, which manifest characteristically with early LV involvement occurring in isolation or preceding RV disease.28 Of note, in the initial report of the DSP gene mutation responsible for ‘Carvajal syndrome’, the cardiac phenotype resembled that of DCM as opposed to the classic ARVC phenotype.29
Phospholamban normally inhibits the sarcoendoplasmic reticulum calcium transport ATPase, and PLN gene mutations cause dysregulated calcium flux, predisposing to prominent arrhythmia and ventricular dysfunction. Mutations in PLN gene are relatively common in the Netherlands, where a founder mutation of PLN (p.Arg14del) is found in up to 15% of probands with ACM. PLN mutation most frequently gives rise to an ACM predominantly affecting the LV, similar to DCM.21
Mutations of the FLNC gene were initially associated with a particular form of skeletal myofibrillar myopathy. The protein product, filamin C, is mainly expressed in skeletal and cardiac muscles and functions at Z-discs and in the subsarcolemmal regions. Truncating mutations of FLNC were subsequently shown to lead to cardiomyopathy, without skeletal muscle involvement. Isolated FLNC-related cardiomyopathy shows an overlapping phenotype with DCM and is characterised by LV systolic dysfunction with mild dilatation and marked fibrosis.19
Diagnosis
In 2020, an international expert report critically reviewed the 2010 Task Force (TF) criteria for the diagnosis of ARVC with the aim to identify areas of potential improvement.13 The 2010 TF guidelines30 exclusively targeted the original RV phenotype features, did not provide diagnostic criteria for the left-sided forms, and lacked tissue characterisation findings by gadolinium-enhanced CMR for detection of myocardial fibrosis, which are determinants of an accurate diagnosis of the LV phenotype.13 22–24
A 2020 international expert consensus document provided upgraded criteria for the diagnosis of ACM, with the introduction of specific diagnostic criteria for the LV phenotype.14 The 2020 international criteria, also known as the ‘Padua criteria’, represents a modern approach to diagnosis of ACM based on research achievements and the clinical and imaging progress over 30 years at the Medical School of the University of Padua. The Padua criteria were reviewed and shared by several international experts, resulting in an international consensus document.
According to the 2020 international criteria, the diagnosis of biventricular and left-dominant variants of ACM is based on the same multiparametric approach recommended by the 2010 TF criteria for classic ARVC, with six categories of criteria which encompass morphofunctional ventricular abnormalities, structural myocardial tissue alterations, electrocardiographic changes of ventricular depolarisation and repolarisation, ventricular arrhythmias, and familial and genetic findings. The criteria considered necessary for the diagnosis of LV disease variants are classified as ‘major’, while those not necessary but contributing to the refinement of LV phenotype characterization as ‘minor’.
The proposed 2020 international criteria by different categories for the diagnosis of RV and LV phenotypes are summarised in table 1.
The ‘Padua criteria’ for diagnosis of arrhythmogenic cardiomyopathy
While based on the 2010 TF criteria myocardial tissue characterisation relied exclusively on the histopathological study by endomyocardial biopsy samples, the 2020 international criteria recommend that structural myocardial abnormalities are investigated by the CMR LGE technique, which allows the identification of replacement of the LV myocardium by fibrofatty scar tissue.13 22–24 31–34
The accuracy of the 2020 diagnostic scoring system for phenotypic diagnosis of left-sided ACM varies according to different disease forms. In case of biventricular ACM, the fulfilment of the diagnostic criteria for the RV phenotype ensures that the concomitant left-sided abnormalities are disease-specific. Hence, the diagnosis of biventricular ACM can be reliably made in the presence of phenotypic features such as morphofunctional and structural abnormalities of both ventricles.
In patients without clinically detectable RV abnormalities, LV phenotypic criteria in isolation do not allow a conclusive diagnosis of ALVC as the morphofunctional and structural LV abnormalities are not sufficiently disease-specific due to the overlap with the phenotypic features of other heart muscle diseases such as DCM, myocarditis or cardiac sarcoidosis.15 Hence, in the presence of consistent LV phenotypic features, demonstration of a pathogenic or likely pathogenic mutation of ACM related genes is mandatory for diagnosis of ALVC.
According to the 2020 international criteria, the diagnosis of each phenotypic variant of ACM requires that at least one criterion from category 1 (ie, morphofunctional ventricular abnormalities) or category 2 (ie, structural myocardial abnormalities), either major or minor, is met. The reason is that ACM is not a channelopathy whose diagnosis relies on demonstration of pathogenic mutations, ECG abnormalities or arrhythmias in the presence of a normal heart, but a cardiomyopathy with distinctive structural ventricular abnormalities, which are an integral part of the phenotype.
Although CMR has become the leading imaging modality for diagnosis of ACM due to its unique capability of identifying fibrofatty myocardial replacement by LGE tissue characterisation, novel and sensitive ultrasound techniques such as strain echocardiography can be useful for detection of early ventricular dysfunction.35 36
In the 2020 international criteria, the scoring system for diagnosis of left-sided phenotypic variants of ACM is substantially unchanged compared with the 2010 TF criteria for diagnosis of ARVC.14 The biventricular variant is diagnosed in patients fulfilling morphofunctional and/or structural abnormalities of both the RV and the LV. The diagnosis is classified as definite when two major, one major and two minor, or four minor criteria of different categories from both ventricles are fulfilled; borderline, one major and one minor, or three minor criteria; and possible, one major or two minor criteria. Of note, ≥1 morphofunctional and/or structural criteria, either major or minor, are needed for each diagnosis. The diagnosis of ALVC, without clinical evidence of RV abnormalities, needs demonstration of ACM-causing gene mutations or familial ACM, in association with consistent LV structural abnormalities (figure 3).

Morphofunctional and structural characteristics of phenotypic variants of arrhythmogenic cardiomyopathy. Note that demonstration of a pathogenic arrhythmogenic cardiomyopathy gene mutation (or familial arrhythmogenic cardiomyopathy) is mandatory for diagnosis of ALVC, in association with consistent structural myocardial abnormalities. ALVC, arrhythmogenic left ventricular cardiomyopathy; ARVC, arrhythmogenic right ventricular (dysplasia/) cardiomyopathy; LV, left ventricle; RV, right ventricle.
Differential diagnosis
The ALVC phenotype can overlap with that of other genetic cardiomyopathies, neuromuscular diseases, or non-genetic disorders such as acquired DCM, myocarditis and sarcoidosis.13 These latter heart muscle disorders represent the most challenging differential diagnoses with ALVC.
Dilated cardiomyopathy
DCM is the most common condition requiring a differential diagnosis with ALVC due to overlapping phenotypic features such as LV systolic dysfunction and myocardial fibrosis. The main discriminant features between the two cardiomyopathies are reported in table 2. Significant differences are identified by CMR imaging study, which consist of the specific LV remodelling pattern, the extent and regional distribution of myocardial fibrosis as evidenced by LGE, and the relation between the amount of LGE and LV systolic dysfunction.16 17 22 The typical ventricular remodelling pattern of ALVC consists of a hypokinetic (or normokinetic) LV, with no or mild cavity dilatation, in comparison with that of DCM characterised by significant LV ventricular dilatation and systolic dysfunction. Tissue characterisation findings by CMR offer the potential to identify and quantify the myocardial LGE/fibrosis. While LGE is detected in <50% of DCM cases, 100% of patients with ALVC with LV systolic dysfunction show the presence of LV LGE.22 The distribution of LGE differs between the two conditions, predominantly affecting the subepicardial inferolateral regions in ALVC versus mid-mural septal segments in DCM (figure 4). In ALVC, the coexistence of fatty myocardial infiltration is often observed on dedicated sequences in the same regions of LGE or in remote LV areas.

Left ventricular remodelling and regional distribution of LGE in ACM-LV phenotype versus DCM-LV phenotype. Comparison between CMR findings in the subgroup of patients with ACM (left) and patients with DCM (right) with overlapping LV phenotypes (ie, LV systolic dysfunction and LV LGE) shows that (1) ACM has lower LV volume and mass, less depressed LVEF (not shown) and greater amount of LV LGE (ie, ‘hypokinetic, non‐dilated and fibrotic left ventricle’) compared with patients with DCM); (2) LV LGE predominately affects the inferolateral segments in ACM and septal segments in DCM; and (3) in ACM the LV LGE appears as a stria pattern that more frequently affects the subepicardial layers, compared with the spot/patchy pattern and the mid-myocardial location of DCM . Modified from Cipriana et al.22 ACM, arrhythmogenic cardiomyopathy; ARVC, arrhythmogenic right ventricular (dysplasia/) cardiomyopathy; CMR, cardiac magnetic resonance; DCM, dilated cardiomyopathy; LGE, late gadolinium enhancement; LV, left ventricular; LVEF, left ventricular ejection fraction.
Differential diagnosis between ALVC and DCM
The amount of LV myocardial fibrosis/LGE is significantly greater in patients with ALVC than in patients with DCM (online supplemental figure 1)17 22 and directly related to the severity of LV systolic dysfunction (online supplemental figure 2)22 because it acts as a pathological marker of myocardial contractile reserve. On the contrary, LV LGE in DCM represents an epiphenomenon and is unrelated to the reduction of the LV systolic function.22
Supplemental material
Supplemental material
In the current guidelines of the European Society of Cardiology, the so-called ‘hypokinetic, non-dilated LV cardiomyopathy’ has been classified as part of the clinical spectrum of DCM and considered as a disease variant with a less expressed phenotype.37 However, there is compelling evidence that this cardiomyopathy remodelling, distinctively characterised by extensive, non-transmural scarring of the LV which scarcely affects the LV global and regional systolic function, fits better with the phenotype of ALVC, suggesting that a sizeable proportion of cardiomyopathies previously diagnosed as morphologically mild expression of DCM should be reclassified as ALVC.16 17 22
The LV systolic dysfunction of ALVC may become more severe in advanced stages, as a result of a disease progression by increasing the extent of LGE which affects multiple segments of the LV free wall and septum, with a more transmural involvement.1 15 Demonstration of an ACM-causing gene mutation in association with a consistent phenotype is mandatory for diagnosis of ALVC.14
Myocarditis
Isolated LV LGE with subepicardial/mid-myocardial distribution (ie, non-ischaemic) is traditionally interpreted as the consequence of a previous myocarditis, although there is increasing evidence that it may reflect a segmental ALVC (figure 5).25 38 39 Similar to ALVC, a postinflammatory scar may induce ECG changes, such as low-amplitude QRS complexes in limb leads, T-wave inversion and ventricular arrhythmias with RBBB. Moreover, because of its focal and non-transmural wall involvement, the myocardial scar may be undetectable by echocardiography.38 39 Non-ischaemic myocardial scar has been reported to act as a myocardial substrate for life-threatening ventricular arrhythmias and cardiac arrest, mostly in young people and athletes. A previous study reported that during a mean 3-year follow-up 22% of athletes with LV scar-related ventricular arrhythmias experienced major arrhythmic events including SCD, compared with none of the control athletes with ventricular arrhythmias in the absence of LV scar.25 The most important clinical implication of detection of a non-ischaemic LV scar is that it cannot be dismissed as a benign sign of a healed remote inflammatory process but deserves proper clinical attention. Affected patients should be evaluated for a family history of cardiomyopathy, clinical history of acute myocarditis, presence of symptoms and the arrhythmogenicity of the myocardial fibrosis. Molecular genetic testing with demonstration of ACM gene mutation is needed in selected non-familial cases for a conclusive differential diagnosis between ALVC and myocarditis.
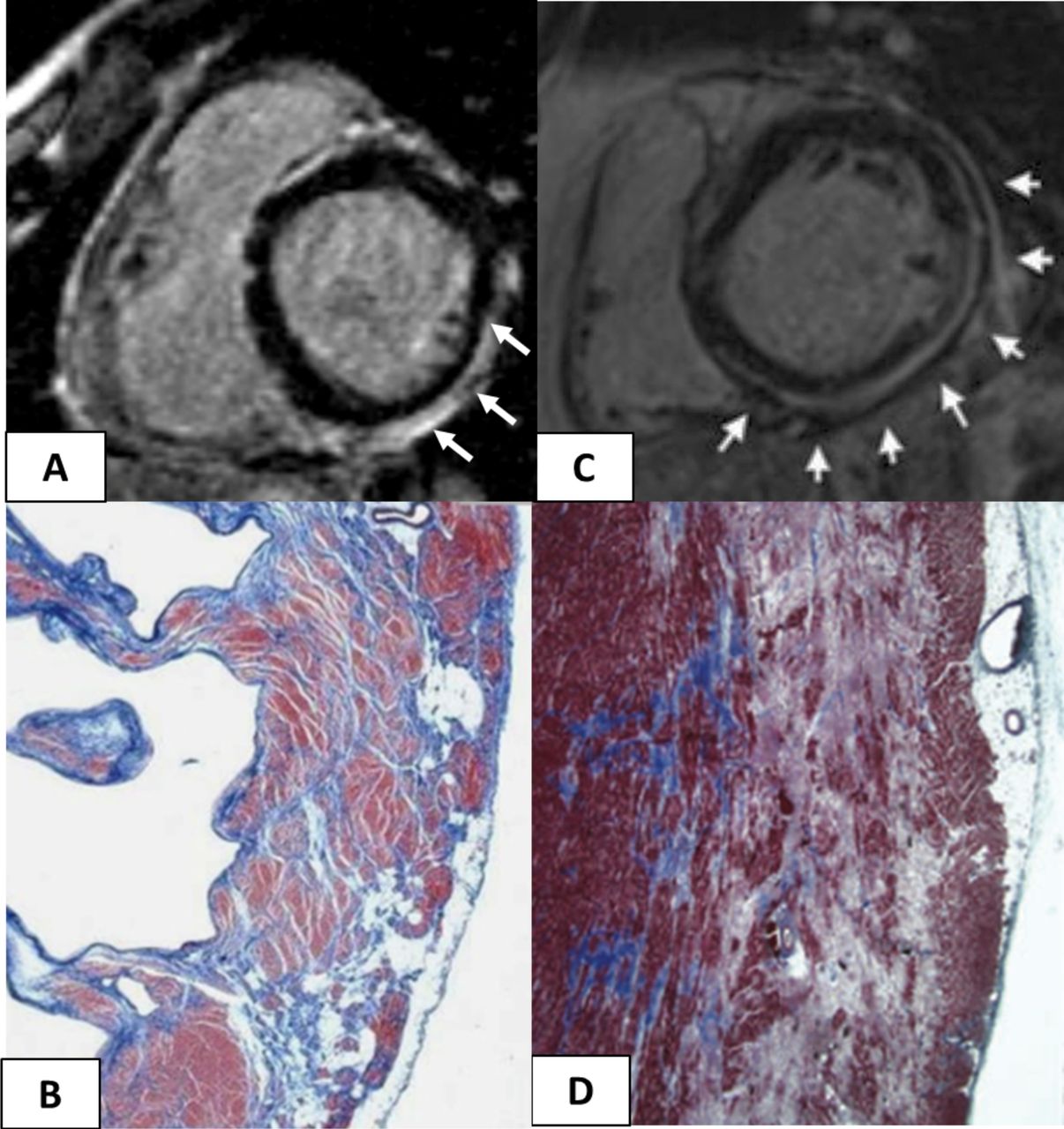
Cardiac magnetic resonance features and histopathological findings of structural changes in myocarditis and ALVC. Myocarditis (A and B): postcontrast T1 inversion recovery sequence in short-axis view showing subepicardial LGE of the inferolateral LV wall (arrows) (A); corresponding panoramic histopathological view of the inferolateral LV wall showing extensive fibrous tissue replacement in the subepicardial layer of the myocardium (B). Desmosomal gene-related ALVC in a sudden cardiac death victim carrying a DSP mutation (C and D): postcontrast T1 inversion recovery sequence in short-axis view showing subepicardial LGE of the inferolateral LV wall (arrows) in a DSP gene mutation carrier (C). Panoramic histopathological view showing fibrofatty myocardial replacement of the outer layer of the inferolateral LV wall (D). Modified from Corrado et al. 13 ALVC, arrhythmogenic left ventricular cardiomyopathy; LGE, late gadolinium enhancement; LV, left ventricle.
Cardiac sarcoidosis
Clinical manifestations common to cardiac sarcoidosis and ALVC include myocardial scarring with dilatation/dysfunction leading to life-threatening ventricular arrhythmias and heart failure. The most important feature distinguishing cardiac sarcoidosis from ALVC is the presence of infrahisian conduction abnormalities such as bundle branch block and atrioventricular block as a consequence of the predilection of granulomatous infiltration for the basal interventricular septum, which is usually absent in ALVC.40 Although definitive diagnosis of sarcoidosis is made by demonstration of granulomatous infiltration on endomyocardial or extracardiac biopsy, non-invasive imaging modalities such as CMR41 and positron emission tomography42 are increasingly used in the clinical setting.
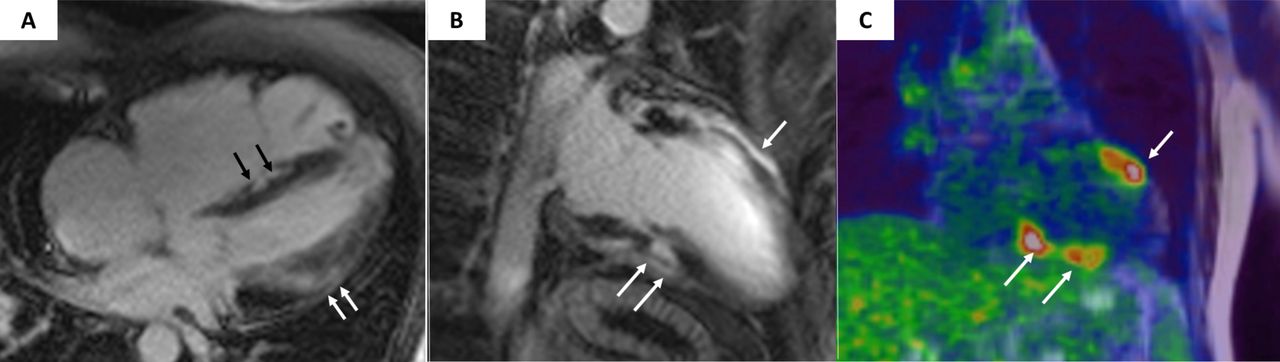
Suggestive of cardiac sarcoidosis are findings of LGE localised in the basal septum and regional fluorodeoxyglucose uptake which indicates active inflammatory lesions (figure 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Magnetic resonance and cardiac positron emission tomography features of cardiac sarcoidosis. Postcontrast T1 inversion recovery sequence in four-chamber view showing LGE of the interventricular septum (black arrows) and subepicardial lateral LV wall (white arrows) (A). Topographic concordance between the epicardial spot of LGE involving the anterior (B, single white arrow) and inferior (B, white arrows) LV regions and the areas of fludeoxyglucose uptake on positron emission tomography (C, white arrows pointing to yellow–red areas). Modified from Corrado et al. 13 LGE, late gadolinium enhancement; LV, left ventricle.
Risk stratification and therapy
The first observational study on the clinical impact of implantable cardioverted defibrillator (ICD) therapy on the natural history of 134 patients with ARVC showed that nearly half of patients had at least one episode of ventricular tachyarrhythmia that required ICD intervention over a mean follow-up period of 3.3 years and 24% experienced ventricular fibrillation/flutter that in all likelihood would have been fatal without termination by the device. On multivariate analysis, independent predictors of ventricular fibrillation/flutter included history of cardiac arrest or ventricular tachycardia with haemodynamic compromise, younger age and left ventricular involvement (evaluated as a reduction of the LV ejection fraction).43 Subsequently, a study aimed to identify the risk factors for long-term prognosis among 130 patients with ARVC showed that during a mean follow-up of 8 years, 24 patients died (21 from cardiovascular death), with an annual mortality rate of 2.3%. Clinical signs of RV failure and LV dysfunction were independently associated with cardiovascular deaths.44 Recent imaging studies reported that LV involvement and LV-dominant presentation (ie, ALVC) predicted a worse prognosis compared with isolated RV disease or normal CMR.45 46
The appropriate management strategy for ALVC is not completely established.47 Restriction of sports activity and prophylactic β-blocker therapy are indicated to prevent ‘adrenergic dependent’ ventricular arrhythmias. Successful mapping/catheter ablation of scar-related sustained ventricular tachycardia (VT)is feasible using an epicardial approach through the pericardial space to reach the re-entry circuit which is located in the outer layer of LV wall.48 Only the implantation of an ICD has been proven to successfully prevent SCD by interrupting potentially lethal ventricular arrhythmias.43 47 According to the 2015 TF consensus documents, three categories of arrhythmic risk (‘high’, ‘moderate’ and ‘low’ risk) are considered for indication to ICD implantation.47 It is noteworthy that the LV systolic function was a key factor for risk stratification. The ‘high-risk’ category (class I recommendation to ICD implantation) comprised patients with a history of cardiac arrest or haemodynamically unstable VT or those with severe ventricular dysfunction, either right (RV fractional area change ≤17% or RV ejection fraction ≤35 %) or left (LV ejection fraction ≤35%). ICD implantation was also indicated (class IIa recommendation) in patients from the ‘intermediate risk’ category with ≥1 ‘major’ risk factors, such as syncope, non-sustained VT or moderate right (RV fractional area change 17%–24% or RV ejection fraction 36%–40%) and/or left (LV ejection fraction 36%–45%) ventricular dysfunction. This reflects the concept that, at variance with DCM, the pathogenesis of life-threatening ventricular arrhythmias in ALVC extends beyond the severe depression of LV systolic function, being strongly related to the large amount of myocardial fibrosis, which is an independent arrhythmogenic risk factor.22 23 Thus, ICD therapy for primary prevention of SCD should be considered earlier in the disease course, when the degree of LV systolic dysfunction is still mild or moderate.13 Subcutaneous ICD has been proven to substantially reduce lead-related complications but maintaining efficacy and should be considered for primary prevention in young patients.49
Patients with advanced ALVC and severe systolic dysfunction are treated with the traditional heart failure therapy.47 Heart transplantation is the final therapeutic option for those patients with either end-stage and unresponsive congestive heart failure or VT/ventricular fibrillation storms refractory to catheter (and surgical) ablation and/or ICD therapy.50
Summary/conclusions
The spectrum of ACM phenotypes is broader than previously believed and besides ARVC includes biventricular variants and ALVC. The 2010 TF criteria lacked specific criteria for diagnosis of left-sided phenotypes, which resulted in underdiagnosis of ACM. The 2020 international diagnostic criteria were provided to fill the gaps of previous TF guidelines, mostly by introduction of a new criteria for the diagnosis of the LV phenotype, mostly based on CMR myocardial tissue characterisation findings which are determinants for detection of ALVC. Clinical studies on large patient cohorts are needed to validate the accuracy and quantitate the impact of the 2020 international criteria in diagnosing the entire spectrum of ACM variants, with particular reference to ALVC.
Future directions of research include comprehensive understanding of the pathogenesis, increased awareness of clinical manifestations and outcome, as well as more accurate risk stratification and effective therapy for ALVC.
Key messages
Arrhythmogenic cardiomyopathy (ACM) is a biventricular muscle disease with left ventricle (LV) involvement which may parallel (biventricular) or exceed (left-dominant/arrhythmogenic left ventricular cardiomyopathy (ALVC)) the severity of the right ventricle (RV) disease.
‘Functional’ imaging evaluation of the LV is insufficient for diagnosis of ALVC, which requires myocardial tissue characterisation by the late gadolinium enhancement technique.
In patients with an ALVC phenotype (and no RV abnormalities), demonstration of a pathogenic mutation in ACM-related genes is mandatory for diagnosis.
Implantable cardioverted defibrillator implantation for primary prevention should be considered in the presence of a large arrhythmogenic ventricular scar, even if the LV systolic function is not severely depressed.
CME credits for Education in Heart
Education in Heart articles are accredited for CME by various providers. To answer the accompanying multiple choice questions (MCQs) and obtain your credits, click on the ‘Take the Test’ link on the online version of the article. The MCQs are hosted on BMJ Learning. All users must complete a one-time registration on BMJ Learning and subsequently log in on every visit using their username and password to access modules and their CME record. Accreditation is only valid for 2 years from the date of publication. Printable CME certificates are available to users that achieve the minimum pass mark.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Both authors gave substantial contributions to conception of the work and drafting the work. Both gave final approval of the version published and agree to be accountable for all aspects of the work.
Funding This work was supported by the registry for Cardio-Cerebro-Vascular Pathology, Veneto Region, Venice, Italy; Ministry of Health grant RFİ\2013İ\02356762 and RFİ\2014İ\00000394; University Research grant BIRD162733, Padua, Italy; PRIN Ministry of Education, University and Research 2015ZLNETW_001, Rome, Italy; and the CARIPARO Foundation, Padua, Italy.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Author note References which include a * are considered to be key references.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.