





Abstract
HYpothesis testing using PHYlogenies (HyPhy) is a scriptable, open-source package for fitting a broad range of evolutionary models to multiple sequence alignments, and for conducting subsequent parameter estimation and hypothesis testing, primarily in the maximum likelihood statistical framework. It has become a popular choice for characterizing various aspects of the evolutionary process: natural selection, evolutionary rates, recombination, and coevolution. The 2.5 release (available from www.hyphy.org) includes a completely re-engineered computational core and analysis library that introduces new classes of evolutionary models and statistical tests, delivers substantial performance and stability enhancements, improves usability, streamlines end-to-end analysis workflows, makes it easier to develop custom analyses, and is mostly backward compatible with previous HyPhy releases.
Introduction
HYpothesis testing using PHYlogenies (HyPhy) is an open-source software package for comparative sequence analysis using stochastic evolutionary models for codon, protein, nucleotide, and other discrete character data. HyPhy is written in C++14 and implements a domain-specific scripting language (HBL, or HyPhy Batch Language) reminiscent in syntax and conventions to JavaScript. HyPhy follows the increasingly common design paradigm of coupling a comprehensive computational engine that implements compute-intensive functionality and essential data types with a rich and extensible library of scripts, plug-ins, or modules that are used to implement models, analyses, and high-level algorithms, for example, R (R Core Team 2013), RevBayes (Höhna et al. 2016), BEAST 2.× (Bouckaert et al. 2019). HyPhy is distributed with a library of HBL modules and prewritten analyses. Since its initial release in 2000 and publication in 2005 (Pond et al. 2005), HyPhy and its companion Datamonkey web application (Weaver et al. 2018) have become integral tools for the bioinformatics community. These resources have collectively generated over 4,500 peer-reviewed citations, with ∼1,000 jobs processed each week by Datamonkey with no charge to the scientific community. Extensions and improvements to HyPhy have been ongoing since its initial release, with active feedback between users and developers producing new features tailored to the specific needs of the research community. Here, we announce the release of HyPhy version 2.5 and document how the software has been packaged for easy use in a variety of settings, optimized for larger data sets, redesigned to follow modern best practices in software design and engineering, and extended to include a broader set of evolutionary models and prepackaged analyses. We strove to maintain maximal backward compatibility with previous releases, with the exception of several obscure features that have been deprecated, and more rigorous runtime error condition checking.
The source code, installation instructions, and documentation for HyPhy are available at github.com/veg/hyphy and hyphy.org, and the accompanying JavaScript result visualization module—at github.com/veg/hyphy-vision. In addition, a public instance of the HyPhy-powered web-application, implementing the most popular analyses, is available at www.datamonkey.org (Weaver et al. 2018).
New Approaches
Part of the Software Ecosystem
Software development practices and usage patterns have decisively shifted away from monolithic packages toward modular and reusable components that must be installed, versioned, and maintained in complex software ecosystems (Johanson and Hasselbring 2018). Recognizing that users of HyPhy vary greatly in their technical proficiency, from biologists unfamiliar with the command-line to bioinformaticians who want to incorporate HyPhy into their own software, we offer multiple ways to interact with the package (see fig. 1), spanning the spectrum from “one-button click” analyses accessed via web applications to complex, custom model development, and pipeline integration.
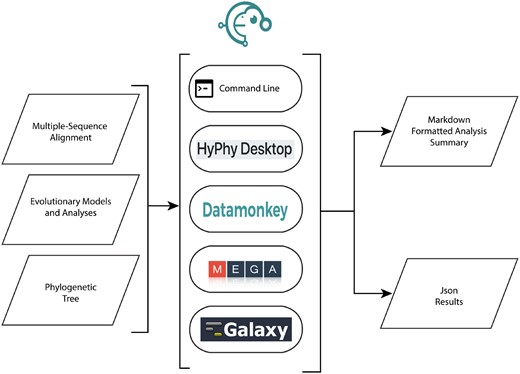
The common use of HyPhy is to fit evolutionary models and perform hypothesis testing on multiple sequence alignments with given phylogenies that are assumed to have been generated with other tools. HyPhy can be run on a desktop, either as a command-line tool for access to its complete functionality, or via a simple Electron-based graphical user interface to access the most popular analyses. Computing cluster use and pipeline integration is facilitated via flexible specification of command line arguments. Several web services, Datamonkey (Weaver et al. 2018), Galaxy (Blankenberg et al. 2010), or MEGA X (Kumar et al. 2018) incorporate HyPhy as a computational engine and provide varying sets of analysis options. Canonical analysis output is accessed in summary/report form via a Markdown document or interactively/programmatically through the detailed JSON report.
Optimized for Larger Data Sets
HyPhy has been optimized extensively to enable analysis of modern large-scale molecular data sets. Core numerical and optimization routines take advantage of vector processing (SIMD), multiprocessing (OpenMP), and distributed systems (MPI) for fine and coarse-grain parallelization as appropriate to a specific analysis. As the majority of popular analyses implemented in HyPhy do not significantly benefit from GPU acceleration compared with tuned multicore CPU optimizations (in our hands at least, also see Izquierdo-Carrasco et al. 2013), we currently do not offer this capability. However with evolving GPU technical characteristics, and refinements open-source libraries supporting gpGPUs for phylogenetics, such as BEAGLE v3 (Ayres et al. 2019), we will continue evaluating this feature. Even when fully accelerated, phylogenetic likelihood calculation for complex models on large data sets remains a bottleneck that additional code tuning cannot fully overcome, for example, even a 100× fold speedup per likelihood function calculation cannot overcome a quadratic dependence of the number of likelihood calculations on the size of the data set. However, we have successfully integrated recent algorithmic advances from the fields of machine learning and natural language processing (Blei et al. 2003) to limit the number of expensive likelihood calculations for certain key applications, namely high-throughput screens for evidence of natural selection along alignments containing thousands of sequences (Murrell et al. 2013). Additional analyses employing these cutting-edge techniques are being actively developed. Finally, we provide high performance computing infrastructure (Weaver et al. 2018) free of charge to the global community, which can be accessed via an internet browser, with the ultimate goal of democratizing access to scientific computing resources (Langmead and Nellore 2018).
Redesigned to Follow Modern Best Practices in Scientific Software
The original HyPhy implementation emphasized convenient writing and execution of single HBL scripts. Command line prompts were used liberally to guide users interactively through selecting the desired analysis and choosing the specific analysis parameters. In addition, the HBL itself was designed to allow sophisticated models to be specified and fit with concise scripts, facilitated by extensive use of a global namespace. However, over the last decade, common use of HyPhy has shifted from one-off analyses toward larger studies which often entail integration into pipelines and web applications. As such, certain key elements of HyPhy have been redesigned to address the requirements of these more modern use cases.
Usage as a typical command-line tool (i.e., an executable name followed by key word arguments) has been added alongside the interactive command line prompt. This change, along with the changes to better conform to standard conventions (e.g., ability to recognize file paths relative to the user’s current working directory and not to the HBL script being called), has allowed for seamless HyPhy integration into pipelines and batch analyses. In addition to being easier to use, HyPhy is also now easier to install, with options to install with bioconda (Grüning et al. 2018) and other package managers like homebrew. Therefore, most users will no longer need to concern themselves with dependencies, environments, and build processes but can obtain HyPhy by simply issuing the command conda install hyphy.
The syntax, semantics, and structure of the standard HBL libraries have also been redesigned and improved, providing a more reliable package that is easier to extend and behaves more similarly to familiar language environments like JavaScript. Examples include controlled vocabulary terms for molecular evolutionary modeling, namespaces for larger library management, basic functional-style programming (e.g., map and filter), JavaDoc inline documentation.
We are also standardizing the way HyPhy analyses operate. A command-line workflow collects required inputs and options from the user, either via keyword arguments, or prompts, executes the analysis, reports progress, and key results to the screen (or file) in MarkDown format (which can also be rendered as formatted report documents), and saves a complete set of results to a JSON file. For the most popular analyses, we have developed a JavaScript interactive visualization library (vision.hyphy.org) that reads the outputted JSON files and renders analysis-appropriate views (tables, charts, alignments, and trees) of the results. JSON files can also be easily parsed and processed in Python, R, or any number of tools for subsequent analysis.
Software Engineering
Software quality control and rigorous testing are expected to be a part of any modern development cycle. However, a recent study by Darriba et al. (2018) suggests that these practices have not been widely adopted in the field of phylogenetics and comparative sequence analysis. For HyPhy2.5, we have implemented extensive automated testing as a part of a continuous integration pipeline, which both expedites development and helps ensure healthy software is delivered. Our suite includes unit tests for over 90% of HBL functions, method tests on all the core analyses, and likelihood testing (https://gitlab.com/testiphy/testiphy), which compares the likelihood values calculated by HyPhy with values calculated by other popular likelihood-based phylogenetic software packages (e.g., MrBayes, IQ-Tree, RaXML) and reports any discrepancies for immediate troubleshooting. For the core, we use C++ development and testing toolkits to perform additional quality controls such as static code analysis, testing for memory leaks and other issues, and instrumented code profiling.
Analyses and Models Supported
The majority of users interact with HyPhy via prewritten scripts to apply published techniques to their data. Key analyses in this class include methods for characterizing the action of natural selection on genes (Murrell et al. 2015), specific sites (Pond and Frost 2005a; Murrell et al. 2012, 2013) in genes, and branches of phylogenetic trees (Smith et al. 2015), comparing selective regimes between sites and branches (Wertheim et al. 2015), screening alignments for evidence of recombination (Pond et al. 2006), inferring networks of coevolving sites (Poon et al. 2007), and performing site-level rate estimation (Spielman and Pond 2018). The list of “stock” analyses is constantly growing to reflect methodological developments by us and other groups in the field.
Using HBL scripting, it is possible to instruct HyPhy to fit very general classes of discrete-state continuous-time Markov substitution models to sequence alignments with fixed phylogenies, perform hypothesis testing via constrained model fitting, perform parametric and nonparametric simulations of sequence alignments to enable modern statistical inferences, infer and sample ancestral sequences. Some of the key features of HyPhy are listed below.
Flexible Model Parameter Specification
Any parameter of the evolutionary model (e.g., synonymous or nonsynonymous evolutionary rate in codon models) can be specified as a free parameter (to be estimated by maximum likelihood), an arbitrary function of other parameters, or a random effect (over sites, branches, partitions, or sites and branches). A model can have an arbitrary number of parameters with varying scopes and properties.
Rate Variation Models
For model parameters that are random effects (e.g., site-level rates drawn from a random distribution), HyPhy supports multiple, arbitrary discrete distributions (including fixed and adaptive [Pond and Frost 2005b] schemes to discretize continuous densities), allows distributions to be independent or conditionally dependent on one another (Pond et al. 2010), and supports spatially auto-correlated rates via hidden Markov models (Felsenstein and Churchill 1996).
Flexible Likelihood Functions
HyPhy can handle analyses combining arbitrary numbers of heterogenous data sets, with model parameters inferred jointly or independently from different components of the data. Each branch in a given phylogeny can have its own evolutionary model. Likelihood calculations can be modified to permit regularized or penalized scoring functions, and convergence criteria can be similarly specified at run time (e.g., a function of changes in parameter estimates instead of the log likelihood value).
Machine Learning Features
HyPhy includes modules for likelihood and Bayesian inference from structured data using stochastic context-free grammars (Poon et al. 2009), Bayesian Graphical Models (Poon et al. 2007), and genetic algorithms (Pond et al. 2006)
Simulation
HyPhy can be used to simulate sequence alignments from any model that it can fit facilitating the analysis of statistical properties of different methods and estimation procedures.
Discussion
Version 2.5 of HyPhy is a near-complete redesign of a popular software package for evolutionary hypothesis testing that should benefit all levels of users, while maintaining maximal compatibility with previously developed analyzes. It incorporates the lessons we have learned over nearly two decades from our own experience and through invaluable comments and feedback from our user community in an effort to make HyPhy maximally useful and minimally painful. This release expands the applicability of the software to larger data sets and more complex evolutionary models, and positions HyPhy to remain a useful part of the analytic toolkit for various fields of computational biology, genomics, and biomedical research.
Acknowledgments
This work was supported in part by grants R01 GM093939 (NIH/NIGMS), R01 AI134384 (NIH/NIAID), T32 GM081057 (NIH/NIEHS), and U01 GM110749 (NIH/NIGMS). S.D.W.F. is supported in part by the Alan Turing Institute via an Engineering and Physical Sciences Research Council grant (EP/510129/1).
References
R Core Team.

{kind=link}