





Abbreviations and acronyms
- 2D
two-dimensional
- 99mTc-DPD
99mTechnetium-3,3-diphosphono- 1,2-propanodi-carboxylic acid
- ACE
angiotensin-converting enzyme
- AF
atrial fibrillation
- AL
amyloid light chain
- AR
aortic regurgitation
- ARB
angiotensin receptor blocker
- ATTR
amyloidosis-transthyretin type
- AV
atrioventricular
- BiVAD
biventricular assist device
- BNP
brain natriuretic peptide
- BPM
Beats per minute
- CCS
Canadian Cardiovascular Society
- CFC
cardiofacialcutaneous
- CHA2DS2-VASc
Congestive Heart failure, hypertension, Age ≥75 (doubled), Diabetes, Stroke (doubled), Vascular disease, Age 65–74, and Sex (female)
- CMR
cardiac magnetic resonance
- CRT
cardiac resynchronization therapy
- CRT-D
cardiac resynchronization therapy-defibrillator
- CRT-P
Cardiac resynchronization therapy with a pacemaker
- CT
computed tomography
- DC
direct current
- DNA
deoxyribonucleic acid
- E/A
ratio of mitral peak velocity of early filling (E) to mitral peak velocity of late filling (A)
- E/e’
ratio of early transmitral flow velocity (E) to early mitral annulus velocity (e’)
- EACTS
European Association for Cardio-Thoracic Surgery
- ECG
electrocardiogram
- EF
ejection fraction
- EPS
electrophysiological study
- ESC
European Society of Cardiology
- FDA
(US) Food and Drug Administration
- FHL1
four and a half LIM domains 1
- HAS-BLED
hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile INR, elderly (>65 years), drugs/alcohol concomitantly
- HCM
hypertrophic cardiomyopathy
- hs-cTnT
high sensitivity cardiac troponin T
- HTS
high throughput sequencing
- ICD
implantable cardioverter defibrillator
- ILR
implantable loop recorder
- INR
international normalized ratio
- IUD
intrauterine device
- LA
left atrium
- LAMP-2
lysosome-associated membrane protein 2
- LBBB
left bundle branch block
- LEOPARD
Lentigines, ECG abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, and sensory-neural Deafness
- LGE
late gadolinium enhancement
- LV
left ventricular
- LVAD
left ventricular assist device
- LVH
left ventricular hypertrophy
- LVOTO
left ventricular outlow tract obstruction
- MADIT-RIT
Multicenter Automatic Defibrillator Implantation Trial—Reduce Inappropriate Therapy
- MAPK
mitogen activated protein kinase
- MELAS
mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes
- MERFF
myoclonic epilepsy with ragged red fibres
- MRA
mineralocorticoid receptor antagonist
- MYBPC3
myosin-binding protein C, cardiac-type
- MYH7
myosin-7 (ß-myosin heavy chain)
- MYL3
myosin light chain 3
- NOAC
new oral anticoagulants
- NSVT
non-sustained ventricular tachycardia
- NT-proBNP
N-terminal pro brain natriuretic peptide
- NYHA
New York Heart Association
- OAC
oral anticoagulants
- o.d.
omni die (every day)
- PC-CMR
phase contrast cardiac magnetic resonance
- PDE5
phosphodiesterase type 5
- PET
positron emission tomography
- PRKAG2
gamma-2 sub-unit of the adenosine monophosphate-activated protein kinase
- RAAS
renin angiotensin aldosterone system
- RV
right ventricular
- SAM
systolic anterior motion
- SCD
sudden cardiac death
- SAA
septal alcohol ablation
- S-ICD™
Subcutaneous lead implantable cardioverter defibrillator
- SPECT
single photon emission computed tomography
- SSFP
steady-state free precession
- SVT
supraventricular tachycardia
- TOE
transoesophageal echocardiography
- TNNI3
troponin I, cardiac muscle
- TNNT2
troponin T, cardiac muscle
- TPM1
tropomyosin alpha-1 chain
- TTE
transthoracic echocardiography
- TTR
transthyretin
- VF
ventricular fibrillation
- VKA
vitamin K antagonist
- VT
ventricular tachycardia
- WHO
World Health Organization
1. Preamble
Guidelines summarize and evaluate all available evidence at the time of the writing process, on a particular issue with the aim of assisting health professionals in selecting the best management strategies for an individual patient, with a given condition, taking into account the impact on outcome, as well as the risk-benefit-ratio of particular diagnostic or therapeutic means. Guidelines and recommendations should help the health professionals to make decisions in their daily practice. However, the final decisions concerning an individual patient must be made by the responsible health professional(s) in consultation with the patient and caregiver as appropriate.
A great number of Guidelines have been issued in recent years by the European Society of Cardiology (ESC) as well as by other societies and organisations. Because of the impact on clinical practice, quality criteria for the development of guidelines have been established in order to make all decisions transparent to the user. The recommendations for formulating and issuing ESC Guidelines can be found on the ESC website (http://www.escardio.org/guidelines-surveys/esc-guidelines/about/Pages/rules-writing.aspx). ESC Guidelines represent the official position of the ESC on a given topic and are regularly updated.
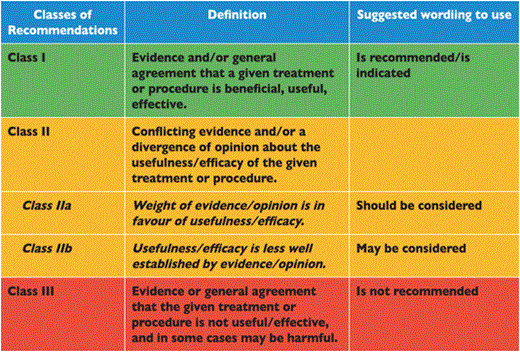
Members of this Task Force were selected by the ESC to represent professionals involved with the medical care of patients with this pathology. Selected experts in the field undertook a comprehensive review of the published evidence for management (including diagnosis, treatment, prevention and rehabilitation) of a given condition according to ESC Committee for Practice Guidelines (CPG) policy. A critical evaluation of diagnostic and therapeutic procedures was performed including assessment of the risk-benefit-ratio. Estimates of expected health outcomes for larger populations were included, where data exist. The level of evidence and the strength of recommendation of particular management options were weighed and graded according to predefined scales, as outlined in Tables 1 and 2.
The experts of the writing and reviewing panels filled in declarations of interest forms which might be perceived as real or potential sources of conflicts of interest. These forms were compiled into one file and can be found on the ESC website (http://www.escardio.org/guidelines). Any changes in declarations of interest that arise during the writing period must be notified to the ESC and updated. The Task Force received its entire financial support from the ESC without any involvement from healthcare industry.
The ESC CPG supervises and coordinates the preparation of new Guidelines produced by Task Forces, expert groups or consensus panels. The Committee is also responsible for the endorsement process of these Guidelines. The ESC Guidelines undergo extensive review by the CPG and external experts. After appropriate revisions it is approved by all the experts involved in the Task Force. The finalized document is approved by the CPG for publication in the European Heart Journal. It was developed after careful consideration of the scientific and medical knowledge and the evidence available at the time of their dating.
The task of developing ESC Guidelines covers not only the integration of the most recent research, but also the creation of educational tools and implementation programmes for the recommendations. To implement the guidelines, condensed pocket guidelines versions, summary slides, booklets with essential messages, summary cards for non-specialists, electronic version for digital applications (smartphones etc) are produced. These versions are abridged and, thus, if needed, one should always refer to the full text version which is freely available on the ESC website. The National Societies of the ESC are encouraged to endorse, translate and implement the ESC Guidelines. Implementation programmes are needed because it has been shown that the outcome of disease may be favourably influenced by the thorough application of clinical recommendations.
Surveys and registries are needed to verify that real-life daily practice is in keeping with what is recommended in the guidelines, thus completing the loop between clinical research, writing of guidelines, disseminating them and implementing them into clinical practice.
Health professionals are encouraged to take the ESC Guidelines fully into account when exercising their clinical judgment as well as in the determination and the implementation of preventive, diagnostic or therapeutic medical strategies. However, the ESC Guidelines do not override in any way whatsoever the individual responsibility of health professionals to make appropriate and accurate decisions in consideration of each patient’s health condition and in consultation with that patient and the patient's caregiver where appropriate and/or necessary. It is also the health professional's responsibility to verify the rules and regulations applicable to drugs and devices at the time of prescription.

2. Introduction
2.1 Definition
Cardiomyopathies are defined by structural and functional abnormalities of the ventricular myocardium that are unexplained by flow-limiting coronary artery disease or abnormal loading conditions.1 Historically, this group of disorders has been subdivided into primary disease, in which the heart is the only involved organ, and secondary forms where the cardiomyopathy is a manifestation of a systemic disorder. These Guidelines adopt a classification system proposed in a recent ESC position statement, in which cardiomyopathies are defined by specific morphological and functional criteria and then grouped into familial/genetic and non-familial/non-genetic subtypes, irrespective of the presence of extra-cardiac disease.1
Hypertrophic cardiomyopathy (HCM) is defined by the presence of increased left ventricular (LV) wall thickness that is not solely explained by abnormal loading conditions.
This definition applies to children and adults and makes no a priori assumptions about aetiology or myocardial pathology. While this approach broadens the scope of the Guidelines and makes some recommendations more complex, it aligns with everyday clinical practice and is more likely to improve diagnostic accuracy and treatment.
2.2 Scope of Guidelines
Uniquely for a common cardiovascular disease, there are very few randomized, controlled, clinical trials in patients with HCM.2 For this reason, the majority of the recommendations in this document are based on observational cohort studies and expert consensus opinion. The aim is to provide healthcare professionals with a practical diagnostic and treatment framework for patients of all ages and, as the majority of patients have a genetic cause for their disease, the Guidelines also consider the implications of a diagnosis for families and provide specific advice on reproduction and contraception.
Adoption of a purely morphological disease definition means that the number of possible aetiologies is considerable, particularly in young children. As it is impractical to provide an exhaustive compendium of all possible causes of HCM, the Guidelines focus on the most common genetic and non-genetic subtypes, but additional references for less common disorders are provided. Similarly, treatment recommendations focus largely on generic management issues but make reference to rare diseases when appropriate.
3. Epidemiology
A number of methodologically diverse studies in North America, Europe, Asia and Africa report a prevalence of unexplained increase in LV thickness in the range of 0.02–0.23% in adults (Web Table 1).3–12 Many show an age-related prevalence, with much lower rates in patients diagnosed under the age of 25 years.9 In paediatric registries, the prevalence of HCM in children is unknown, but population-based studies report an annual incidence of 0.3 to 0.5 per 100,00013,14 (Web Table 1). While HCM is most frequently transmitted as an autosomal-dominant trait (see section 6: Genetic testing and family screening) most studies report a small male preponderance (Web Table 1). This finding remains unexplained but might reflect bias in screening strategies as well as genetic and hormonal modifiers. The prevalence of HCM in different racial groups is similar.3–12
4. Aetiology
In up to 60% of adolescents and adults with HCM, the disease is an autosomal dominant trait caused by mutations in cardiac sarcomere protein genes.15–19
Five to ten percent of adult cases are caused by other genetic disorders including inherited metabolic and neuromuscular diseases, chromosome abnormalities and genetic syndromes (Figure 1; Web Tables 2 and 3).20,21 Some patients have non-genetic disorders that mimic genetic forms of the disease, for example, senile (TTR) and (AL) amyloidosis.22,23
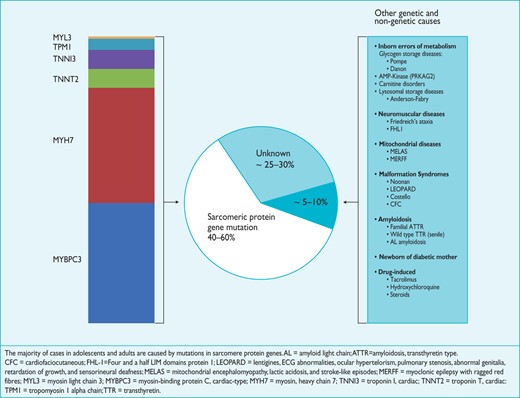
Diverse aetiology of hypertrophic cardiomyopathy.
4.1 Sarcomere protein gene mutations
Mutations in the genes encoding beta-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) account for the majority of cases; less commonly affected genes include cardiac troponin I and T (TNNI3, TNNT2), tropomyosin alpha-1 chain (TPM1) and myosin light chain 3 (MYL3). In general, patients with a sarcomere protein mutation present earlier and report a higher prevalence of family history of HCM and sudden cardiac death (SCD) than those without a mutation.19,24 They also tend to have more severe hypertrophy, microvascular dysfunction and myocardial fibrosis.25 Several studies have suggested that some sarcomeric protein mutations are associated with a poorer prognosis than others, but these observations are based on small numbers of affected individuals, are sometimes inconsistent between studies, and are limited by the rarity of individual mutations.26–32 This situation should improve as better data are collected on individual mutations in international databases such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). Multiple sarcomeric protein mutations are present in up to 5% of individuals and tend to present earlier with a more severe phenotype.33–35
4.2 Metabolic disorders
Many inherited metabolic diseases are associated with LV hypertrophy. Most are inherited as autosomal recessive traits, but a few are X-linked (Figure 1;Web Table 3).21 The most common metabolic disorders in adults with HCM are Anderson-Fabry disease, with a prevalence of around 0.5–1% in patients older than 35–40 years,36 and disease caused by mutations in the gene encoding the γ2 sub-unit of the adenosine monophosphate-activated protein kinase (PRKAG2), with a prevalence of approximately 1%.37 The reported prevalence of lysosome-associated membrane protein 2 (LAMP-2) mutations that cause Danon disease ranges from 0.7–2.7%.38 Although still rare, metabolic disorders account for a greater proportion of HCM in children and adolescents.
4.3 Mitochondrial cardiomyopathies
Primary mitochondrial disorders are caused by mutations in nuclear or mitochondrial DNA that are transmitted as autosomal dominant, autosomal recessive, X-linked and maternally inherited traits.39 The most frequent are those caused by mutations in genes that code for the respiratory chain protein complexes (Web Table 3).21 The clinical presentation of mitochondrial disease typically varies in age at onset and in the range and severity of organ involvement.
4.4 Neuromuscular disease
With the exception of Friedreich's ataxia,40,41 HCM is a rare manifestation of neuromuscular disease (Figure 1; Web Table 3).21 It is reported in some muscular dystrophies and congenital skeletal myopathies (e.g. nemaline myopathy)42 (Web Table 3)21 and in association with muscle weakness and contractures caused by mutations in the four-and-half LIM domain-1 (FHL-1) gene.43 Desmin gene mutations typically cause dilated and restrictive cardiomyopathies, but can present with HCM and atrioventricular (AV) block.44
4.5 Malformation syndromes
Several malformation syndromes are associated with HCM (Web table 3). The most common are those caused by mutations in genes that code for proteins of the Ras/mitogen activated protein kinase (MAPK) pathway including Noonan,45 LEOPARD (Lentigines, ECG abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, and sensorineural Deafness)46,47 and Costello syndromes.48 Most are diagnosed in childhood, but some milder forms (particularly Noonan syndrome) escape early detection and are identified later in life.
4.6 Infiltrative disease/inflammation
Cardiac amyloidosis results in a progressive increase in the thickness of the left and right ventricular myocardium, interatrial septum and AV valves.49 Light chain (AL) and hereditary transthyretin (TTR)-related amyloidoses can affect the heart in isolation or with multi-organ involvement, whereas wild type (senile) TTR amyloidosis predominantly affects the heart and the carpal tunnel ligament. Myocardial oedema and cellular infiltration in acute myocarditis can mimic HCM, but this is usually a transient phenomenon, accompanied by other clinical and laboratory findings suggestive of the diagnosis.50,51
4.7 Endocrine disorders
Transient ventricular hypertrophy is seen in infants of mothers with diabetes, even after good diabetic control during pregnancy.52 In adults, left ventricular hypertrophy (LVH) is reported in association with phaeochromocytoma53 and acromegaly,54 but treatment of the underlying endocrine disorder usually results in resolution of hypertrophy.
4.8 Drugs
Chronic use of some drugs, including anabolic steroids, tacrolimus and hydroxychloroquine, can cause LVH although they rarely result in a left ventricular wall thickness ≥1.5 cm.55–57
5. Diagnosis
The diagnosis of HCM rests on the detection of increased LV wall thickness by any imaging modality, but the disease phenotype also includes myocardial fibrosis, morphologic abnormalities of the mitral valve apparatus, abnormal coronary microcirculatory function and electrocardiographic abnormalities. Due to the diverse aetiology of the disease, detection of increased LV wall thickness that is unexplained by loading conditions should prompt a systematic search for its underlying cause. In many patients, this work-up should include specialized laboratory testing and, in some circumstances, genetic analysis (Figure 2).
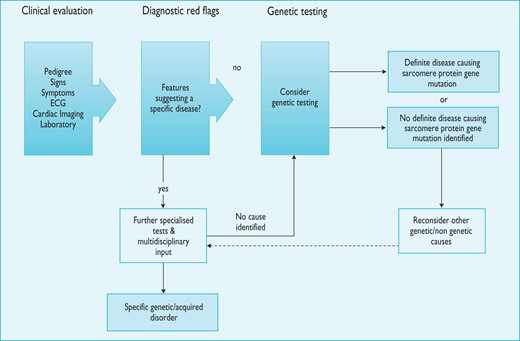
Schematic summarising the general approach to the diagnosis of hypertrophic cardiomyopathy. Notes: 1. Counselling is essential before and after testing for genetic disease. 2. Genetic testing is recommended in patients fulfilling diagnostic criteria for HCM to enable cascade genetic screening of their relatives. 3. For recommendations on individual investigations see relevant sections. ECG = electrocardiogram.
5.1 Diagnostic criteria
5.1.1 Adults
In an adult, HCM is defined by a wall thickness ≥15 mm in one or more LV myocardial segments—as measured by any imaging technique (echocardiography, cardiac magnetic resonance imaging (CMR) or computed tomography (CT))—that is not explained solely by loading conditions.
Genetic and non-genetic disorders can present with lesser degrees of wall thickening (13–14 mm); in these cases, the diagnosis of HCM requires evaluation of other features including family history, non-cardiac symptoms and signs, electrocardiogram (ECG) abnormalities, laboratory tests and multi-modality cardiac imaging.
Common diagnostic challenges include the following:
Presentation in the late phase of the disease with a dilated and/or hypokinetic left ventricle and LV wall thinning (see section 8.2).
Physiological hypertrophy caused by intense athletic training (see section 12.1).
Patients with co-existent pathologies (see section 12.2 on hypertension and section 12.4 on diagnosis and management of valve disease)
Isolated basal septal hypertrophy in elderly people (see section 12.3).
5.1.2 Children
As in adults, the diagnosis of HCM requires an LV wall thickness more than two standard deviations greater than the predicted mean (z-score >2, where a z-score is defined as the number of standard deviations from the population mean).58
5.1.3 Relatives
The clinical diagnosis of HCM in first-degree relatives of patients with unequivocal disease (LVH ≥15 mm) is based on the presence of otherwise unexplained increased LV wall thickness ≥13 mm in one or more LV myocardial segments, as measured using any cardiac imaging technique [echocardiography, cardiac magnetic resonance (CMR) or CT].
In families with genetic forms of HCM, mutation carriers can have non-diagnostic morphological abnormalities that are sometimes associated with abnormal ECG findings. While the specificity of such abnormalities is low, in the context of familial disease they can represent early or mild expression of the disease, and the presence of multiple features increases the accuracy for predicting disease in genotyped populations.59–61 In general, the presence of any abnormality [for example, abnormal Doppler myocardial imaging and strain,62–64 incomplete systolic anterior motion (SAM) or elongation of the mitral valve leaflet(s) and abnormal papillary muscles], particularly in the presence of an abnormal ECG, increases the probability of disease in relatives.59,65,66
5.2 History and physical examination
Age is one of the most important factors to take into account when considering the possible causes for HCM. For example, inherited metabolic disorders and congenital dysmorphic syndromes are much more common in neonates and infants than in older children or adults, whereas wild-type TTR-related amyloidosis is a disease mostly of men over the age of 65 years.
Construction of a three- to four-generation family pedigree helps to confirm a genetic origin of disease and identifies other family members that are at risk of disease development. Specific features to note in the family history include sudden cardiac deaths, unexplained heart failure, cardiac transplantation, pacemaker and defibrillator implants, and evidence for systemic disease (stroke at a young age, skeletal muscle weakness, renal dysfunction, diabetes, deafness, etc.). Pedigree analysis can also determine the likely mode of inheritance. Most genetic forms of HCM are autosomal-dominant (Web Table 2) and are therefore characterized by the presence of affected individuals in every generation, with transmission from parents of either sex (including male to male) and a 50% risk to offspring. X-linked inheritance should be suspected if males are the only or most severely affected individuals and there is no male-to-male transmission. Autosomal recessive inheritance, the least common pattern, is likely when both parents of the proband are unaffected and consanguineous. When women—but not men—transmit the disease to children of either sex, mitochondrial DNA mutations should be considered.
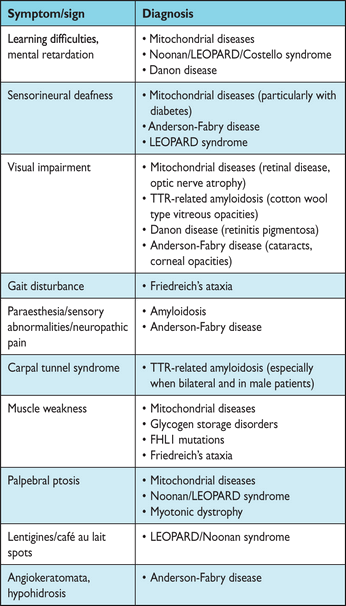
Many individuals with HCM complain of few, if any, symptoms. In such cases the diagnosis can be incidental or the result of screening. Some patients experience angina, dyspnoea, palpitations and syncope (see section 8: Assessment of symptoms). A number of non-cardiac symptoms act as pointers for specific diagnoses (Table 3).67 Similarly, general physical examination can provide diagnostic clues in patients with syndromic or metabolic causes of HCM. Paradoxically, cardiovascular examination is often normal but, in patients with LV outflow tract obstruction (LVOTO), a number of typical features may be identified including a rapid up-and-down stroke to the arterial pulse and an ejection systolic murmur at the left sternal edge that radiates to the right upper sternal edge and apex. The intensity of the murmur is increased by manoeuvres that reduce ventricular preload or afterload, such as standing up from the squatting position and forceful attempted exhalation against a closed airway (Valsalva manoeuvre). Most patients with LVOTO also have signs of mitral regurgitation.
Examples of signs and symptoms suggestive of specific diagnoses (modified from Rapezzi et al.67)
 |
|
FHL1 = four and a half LIM domains 1; LEOPARD = lentigines, ECG abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness; TTR = transthyretin
Examples of signs and symptoms suggestive of specific diagnoses (modified from Rapezzi et al.67)
|
|
FHL1 = four and a half LIM domains 1; LEOPARD = lentigines, ECG abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness; TTR = transthyretin
5.3 Resting and ambulatory electrocardiography
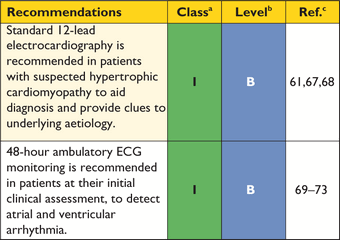
The standard 12-lead ECG can be normal at presentation (6% of patients in referral cohort studies) but generally shows a variable combination of LVH, ST- and T-wave abnormalities, and pathological Q-waves.68 When interpreted in conjunction with findings on echocardiography and CMR imaging, features that would normally indicate other conditions, such as myocardial ischaemia or infarction, can—with age at diagnosis, inheritance pattern and associated clinical features—suggest an underlying diagnosis or provide clues to the distribution of hypertrophy and myocardial scar (Table 4). For this reason, the ECG is recommended at the first clinic visit in all individuals with known or suspected HCM and should be repeated whenever there is a change in symptoms in patients with an established diagnosis. The ECG is also a sensitive—though non-specific—early marker of disease in relatives.61
The frequency of arrhythmias detected during ambulatory electrocardiographic monitoring is age-related. Asymptomatic non-sustained ventricular tachycardia (NSVT), at a rate between 120 and 200 beats per minute (BPM), occurs in 25% of adults with HCM.69,70 Paroxysmal supraventricular arrhythmias occur during ambulatory electrocardiographic monitoring in up to 38% of patients.70 Ambulatory ECG monitoring is recommended at the initial clinical assessment to assess the risk of sudden cardiac death (section 9.5: Sudden cardiac death) and stroke (section 9.4: Atrial tachyarrhythmia).
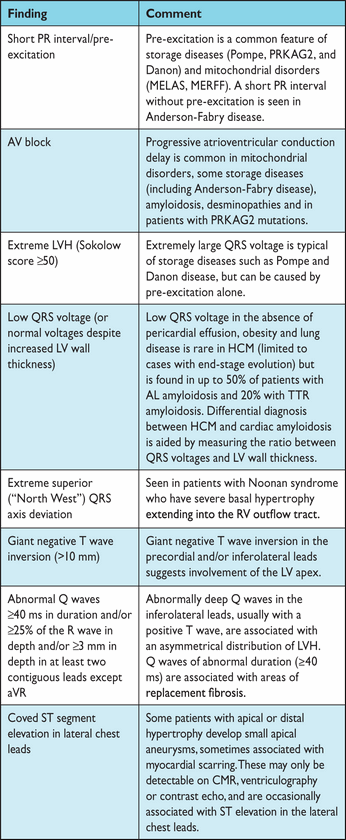
Electrocardiographic abnormalities suggesting specific diagnoses or morphological variants67
 |
|
AV = atrioventricular; AL = amyloid light chain; CMR = cardiac magnetic resonance; HCM = hypertrophic cardiomyopathy; LV = left ventricular; LVH = left ventricular hypertrophy; MELAS = mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERFF = myoclonic epilepsy with ragged red fibres; PRKAG2 = gamma-2 subunit of the adenosine monophosphate-activated protein kinase; RV = right ventricular; TTR = transthyretin.
Electrocardiographic abnormalities suggesting specific diagnoses or morphological variants67
|
|
AV = atrioventricular; AL = amyloid light chain; CMR = cardiac magnetic resonance; HCM = hypertrophic cardiomyopathy; LV = left ventricular; LVH = left ventricular hypertrophy; MELAS = mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERFF = myoclonic epilepsy with ragged red fibres; PRKAG2 = gamma-2 subunit of the adenosine monophosphate-activated protein kinase; RV = right ventricular; TTR = transthyretin.
Recommendations on electrocardiography
 |
|
ECG = electrocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on electrocardiography
|
|
ECG = electrocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
5.4 Echocardiography
Echocardiography is central to the diagnosis and monitoring of HCM. In most patients, hypertrophy preferentially involves the interventricular septum in the basal LV segments but often extends into the lateral wall, the posterior septum and LV apex.74 As increased ventricular wall thickness can be found at any location (including the right ventricle), the presence, distribution and severity of hypertrophy should be documented using a standardized protocol for cross-sectional imaging from several projections. Correct orientation and beam alignment along orthogonal planes are essential to avoid oblique sections and over-estimation of wall thickness. Measurements of LV wall thickness should be performed at end-diastole, preferably in short-axis views. M-mode measurements in the parasternal long axis projection should be avoided if possible, to prevent over-estimation of septal thickness by oblique cuts. A standardized approach to myocardial segmentation and nomenclature should be followed for all imaging modalities.75
5.4.1 Assessment of left ventricular wall thickness
There are a number of echocardiographic indices that provide a semi-quantitative score of LVH, but for diagnostic purposes the single most relevant parameter is the maximum LV wall thickness at any level.
In patients with known or suspected HCM it is essential that all LV segments from base to apex be examined, ensuring that the wall thickness is recorded at mitral, mid-LV and apical levels.
Accurate assessment of LV wall thickness can be challenging when hypertrophy is confined to one or two segments, particularly in the anterolateral wall or the LV apex.74,76–80 In such cases, extra care is needed during imaging (e.g. transducer angulation to avoid problems related to lateral resolution and foreshortening). Similarly, meticulous imaging of the apex by parasternal and multiple apical views is required to detect apical HCM. If a segment is not visualized adequately, LV opacification—using ultrasound contrast agents and/or CMR—should be considered.81
5.4.2 Associated abnormalities of the mitral valve and left ventricular outflow tract
Approximately one-third of patients have resting SAM of the mitral valve leaflets that results in obstruction to the LV outflow tract, while another third have latent obstruction only during manoeuvres that change loading conditions and LV contractility (see 5.4.3: Assessment of latent obstruction).82–85 Other morphological features that contribute to LVOTO include papillary muscle abnormalities (hypertrophy, anterior and internal displacement, direct insertion into the anterior mitral valve leaflet) and mitral leaflet abnormalities such as elongation or accessory tissue.78,86–90 Although dynamic LVOTO is common in patients with HCM, it also occurs in other circumstances, such as calcification of the posterior mitral annulus, hypertension, hypovolaemia and hypercontractile states.
By convention, LVOTO is defined as an instantaneous peak Doppler LV outflow tract pressure gradient ≥30 mm Hg at rest or during physiological provocation such as Valsalva manoeuvre, standing and exercise. A gradient of ≥50 mm Hg is usually considered to be the threshold at which LVOTO becomes haemodynamically important. This concept comes from studies that demonstrate progressive impedance to flow above this value.78
When a gradient is detected in the LV cavity, it is important to systematically exclude obstruction that is unrelated to SAM, including sub-aortic membranes, mitral valve leaflet abnormalities and mid-cavity obstruction, particularly when interventions to relieve LV outflow obstruction are contemplated.
Systematic two-dimensional (2D) and Doppler echocardiography is usually sufficient to determine the mechanism and severity of LVOTO but, when non-invasive images are poor, transoesophageal echocardiography (TOE) or invasive pressure measurements combined with CMR may be considered in selected patients.
Systolic anterior motion of the mitral valve nearly always results in failure of normal leaflet coaptation and mitral regurgitation, which is typically mid-to-late systolic and inferolaterally oriented; measurement of the velocity and timing of the mitral jet helps to differentiate it from LV outflow tract turbulence. SAM-related mitral regurgitation is inherently dynamic in nature and its severity varies with the degree of LVOTO.78,91,92
The presence of a central- or anteriorly directed jet of mitral regurgitationshould raise suspicion of an intrinsic mitral valve abnormality and prompt further assessment with TOE if necessary.
5.4.3 Assessment of latent obstruction
Identification of LVOTO is important in the management of symptoms and assessment of sudden cardiac death risk (see section 9.5: Sudden cardiac death). 2D and Doppler echocardiography during a Valsalva manoeuvre in the sitting and semi-supine position—and then on standing if no gradient is provoked—is recommended in all patients (Figure 3).78,93 Exercise stress echocardiography is recommended in symptomatic patients if bedside manoeuvres fail to induce LVOTO ≥50 mm Hg. Pharmacological provocation with dobutamine is not recommended, as it is not physiological and can be poorly tolerated. Similarly, nitrates do not reproduce exercise-induced gradients and should be reserved for patients who cannot perform physiologically stressful procedures.94 There is some evidence that post-prandial gradients are higher than those performed in the fasting state and pre-treatment with ß-blockers often reduces the incidence and severity of exercise-induced LV outflow tract gradients.95 Since there are relatively few data comparing stress echocardiography protocols,93,95–98 laboratories should develop and validate their own and ensure that staff are properly trained in the procedure.
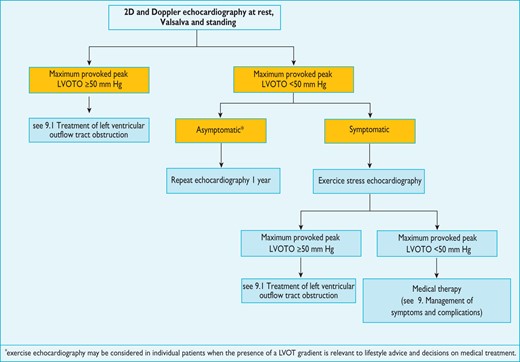
Protocol for the assessment and treatment of left ventricular outflow tract obstruction.
In asymptomatic patients, bedside provocation manoeuvres are useful in risk stratification (see section 9.5: Sudden cardiac death) but routine exercise stress echocardiography in this situation has not been prospectively evaluated and should only be considered in selected patients when the presence of a LVOT gradient is relevant to lifestyle advice and decisions on medical treatment.
5.4.4 Left atrial enlargement
The left atrium (LA) is often enlarged, and its size provides important prognostic information.72,73,99 Although most published studies use anteroposterior LA diameter,100 comparable findings using LA volume indexed to body surface area are reported.101,102 The cause of LA enlargement is multifactorial, but the most common mechanisms are SAM-related mitral regurgitation and elevated LV filling pressures.
5.4.5 Assessment of diastolic function
Patients with HCM often have diastolic dysfunction and the assessment of LV filling pressures is helpful in the evaluation of symptoms and disease staging. Doppler echocardiographic parameters are sensitive measures of diastolic function, but are influenced by loading conditions, heart rate and age, and there is no single echocardiographic parameter that can be used as a diagnostic hallmark of LV diastolic dysfunction.103 Therefore, a comprehensive evaluation of diastolic function—including Doppler myocardial imaging, pulmonary vein flow velocities, pulmonary artery systolic pressure and LA size—is recommended as part of the routine assessment of HCM.103 Patients with a restrictive LV filling pattern [ratio of mitral peak velocity of early filling (E) to mitral peak velocity of late filling (A) ≥2; E-wave deceleration time ≤150 ms] may be at higher risk for adverse outcome, even with a preserved ejection fraction (EF).104,105 Data on the relationship between non-invasive Doppler myocardial imaging-derived estimates of LV filling pressure and invasive pressure studies are contradictory,106 but some studies show correlation between an elevated ratio of early transmitral flow velocity (E) to early mitral annulus velocity (e') >12–15 and raised LV end-diastolic pressure, exercise capacity and prognosis.107,108
5.4.6 Systolic function
Radial contractile function (EF or fractional shortening) is typically normal or increased in patients with HCM. However, EF is a poor measure of LV systolic performance when hypertrophy is present.109 Myocardial longitudinal velocities and deformation parameters (strain and strain rate), derived from Doppler myocardial imaging or speckle tracking techniques, are often reduced despite a normal EF and may be abnormal before the development of increased wall thickness in genetically affected relatives. Myocardial longitudinal deformation is typically reduced at the site of hypertrophy.110
5.4.7 Value of echocardiography in differential diagnosis
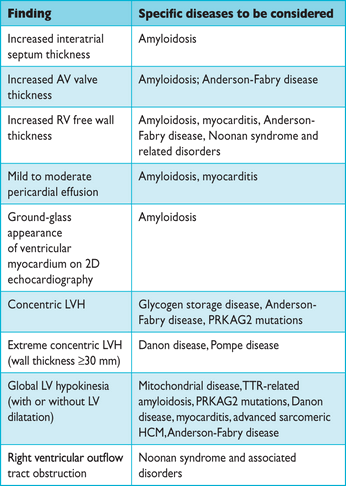
A number of echocardiographic features can point to a specific diagnosis (Table 5).67 Concentric hypertrophy is more common in metabolic and infiltrative disorders and biventricular hypertrophy and obstruction to the outflow of both ventricles is frequent in Noonan syndrome and associated disorders. Clues that suggest myocardial storage disease or infiltration include sparkling or granular myocardial texture, small pericardial effusion, thickening of the interatrial septum, nodular thickening of the aortic valve, and mildly reduced EF with restrictive physiology.
Echocardiographic features that suggest specific aetiologies (modified from Rapezzi et al.67)
 |
|
2D= two-dimensional; AV = atrioventricular; HCM = hypertrophic cardiomyopathy; LV = left ventricular; LVH = left ventricular hypertrophy; PRKAG2 = gamma-2 subunit of the adenosine monophosphate-activated protein kinase; RV = right ventricle; TTR = transthyretin.
Echocardiographic features that suggest specific aetiologies (modified from Rapezzi et al.67)
|
|
2D= two-dimensional; AV = atrioventricular; HCM = hypertrophic cardiomyopathy; LV = left ventricular; LVH = left ventricular hypertrophy; PRKAG2 = gamma-2 subunit of the adenosine monophosphate-activated protein kinase; RV = right ventricle; TTR = transthyretin.
5.4.8 Contrast echocardiography
Apical hypertrophy may be overlooked due to near-field artefacts. Poor visualization of the lateral LV wall may also obscure localized hypertrophy. In cases of doubt, intravenous ultrasound contrast agents should be used to outline the endocardium.81
In all patients undergoing septal alcohol ablation (SAA), intracoronary contrast echocardiography is recommended to ensure correct localization of alcohol(see section 9.1.3.2: Septal alcohol ablation).111–113
5.4.9 Transoesophageal echocardiography
Transoesophageal echocardiography should be considered in patients with poor transthoracic echo windows, as an alternative or complementary investigation to CMR. It is particularly useful in patients with LVOTO if the mechanism is unclear, when assessing the mitral valve apparatus before a septal reduction procedure, and when severe mitral regurgitation caused by intrinsic valve abnormalities is suspected.114–117 In patients undergoing septal myectomy, perioperative TOE should be used to guide the surgical strategy and to detect surgical complications (ventricular septal defect and aortic regurgitation (AR)) and residual LVOTO.116–118 Rarely, TOE with intracoronary contrast injection of the candidate septal perforator arteries is necessary to guide septal alcohol ablation when transthoracic windows are insufficient to visualize contrast within the myocardium.
Recommendations for transthoracic echocardiographic evaluation in hypertrophic cardiomyopathy
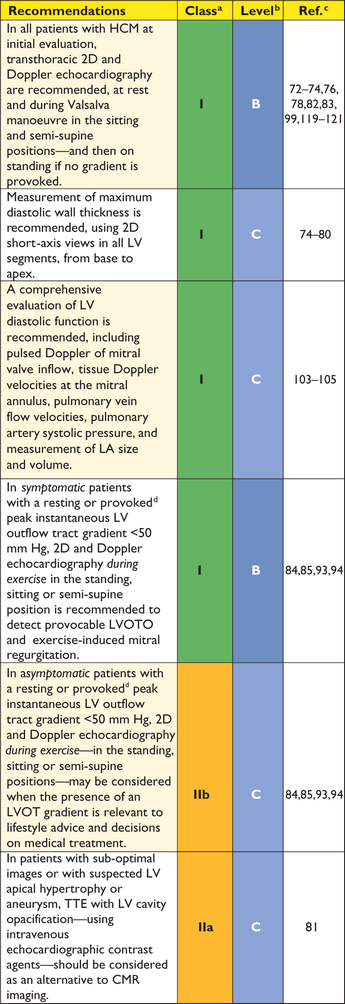 |
 |
|
|
2D = two-dimensional; CMR = cardiac magnetic resonance; LA = left atrium; LV = left ventricle; LVOTO = left ventricular outflow tract obstruction; SAA = septal alcohol ablation; TTE = transthoracic
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dProvocation with Valsalva, standing or oral nitrate.
Recommendations for transthoracic echocardiographic evaluation in hypertrophic cardiomyopathy
|
|
|
|
2D = two-dimensional; CMR = cardiac magnetic resonance; LA = left atrium; LV = left ventricle; LVOTO = left ventricular outflow tract obstruction; SAA = septal alcohol ablation; TTE = transthoracic
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dProvocation with Valsalva, standing or oral nitrate.
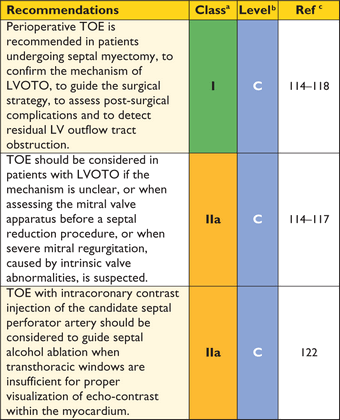
Recommendations on transoesophageal echocardiography
 |
|
LVOTO = left ventricular outflow tract obstruction; TOE = transoesophageal echocardiography.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on transoesophageal echocardiography
|
|
LVOTO = left ventricular outflow tract obstruction; TOE = transoesophageal echocardiography.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
5.5 Cardiovascular magnetic resonance imaging
Cardiovascular magnetic resonance imaging embraces several modalities that provide detailed information on cardiac morphology, ventricular function and myocardial tissue characteristics123. Cardiovascular magnetic resonance evaluation of patients with known or suspected HCM should be in line with current ESC recommendations (http://www.escardio.org/communities/EACVI) and should be performed and interpreted by teams experienced in cardiac imaging and in the evaluation of heart muscle disease.
5.5.1 Assessment of ventricular morphology and function
CMR should be considered in patients with HCM at their baseline assessment if local resources and expertise permit.
In patients with good echocardiographic images, CMR provides similar information on ventricular function and morphology,124,125 but it is helpful in establishing the diagnosis of HCM in patients with poor acoustic windows or when some LV regions are poorly visualized—such as the anterolateral wall, the LV apex and the right ventricle.126,127 As in 2D echocardiography, over-estimation of wall thickness can result from oblique sections (particularly at the LV apex) or from inclusion of paraseptal structures such as the moderator band or false tendons. Over-estimation of wall thickness is also possible in spoiled gradient echo images and so steady-state free precession (SSFP) cine sequences are preferred. Cardiovascular magnetic resonance imaging is superior to transthoracic echocardiography (TTE) in the measurement of LV mass, but LV mass itself correlates weakly with maximal wall thickness and can be normal in patients with asymmetric HCM, especially when it involves less than two LV segments.124,128 Cardiovascular magnetic resonance imaging is superior to standard 2D echocardiography in the detection of LV apical and anterolateral hypertrophy, aneurysms129 and thrombi,130 and is more sensitive in the detection of subtle markers of disease, such as myocardial crypts and papillary muscle abnormalities in patients with sarcomeric protein gene mutations.131–133
Phase velocity flow mapping sequences can be used to determine the peak velocity of blood flow through the LV outflow tract in patients with LVOTO, but proper alignment of the imaging plane, to obtain the highest flow velocities is time-consuming and prone to error. Intravoxel dephasing and signal loss due to phase offset errors, also make the accurate quantification of turbulent flow difficult and LV outflow gradients can only be measured at rest. For these reasons, Doppler echocardiography is the modality of choice for quantification of LVOTO. Similarly, while mitral inflow velocities and pulmonary vein flow derived from phase contrast CMR (PC-CMR) provide highly reproducible and accurate data in experienced hands, echocardiography is the preferred method for assessment of diastolic function in routine practice.103
In selected cases where echocardiographic images are suboptimal, CMR is helpful in pre-operative planning for surgical myectomy, particularly in patients with multi-level LV obstruction (LV outflow tract and mid-cavity) and in patients with right ventricular (RV) outflow tract abnormalities. CMR can also quantify the amount of tissue necrosis induced by septal alcohol ablation, as well as the location of scarring and the regression of LV mass following the procedure.134,135
5.5.2 Myocardial fibrosis
By using the intrinsic magnetic properties of different tissues and the distribution of gadolinium-based contrast agents, CMR can be used to detect expansion of the myocardial interstitium caused by fibrosis. Late gadolinium enhancement (LGE) is present in 65% of patients (range 33–84%), typically in a patchy mid-wall pattern in areas of hypertrophy and at the anterior and posterior RV insertion points.136 Late gadolinium enhancement is unusual in non-hypertrophied segments except in advanced stages of disease, when full-thickness LGE in association with wall thinning is common.136 Late gadolinium enhancement may be associated with increased myocardial stiffness and adverse LV remodelling and the extent of LGE is associated with a higher incidence of regional wall motion abnormalities. Late gadolinium enhancement varies substantially with the quantification method used and the 2-standard deviation technique is the only one validated against necropsy.137
Assessment of LGE before invasive treatment of LVOTO may be useful in selecting the most appropriate therapy by assessing the degree of septal fibrosis (see section 9.1).
5.5.3 Late Gadolinium Enhancement and Prognosis
The association between LGE and long-term outcomes has been examined in six studies,138–143 four of which are included in a meta-analysis (Web Table 4).144 All published studies are limited by selection and referral bias, incomplete risk assessment and differences in scanning protocols and LGE quantification. The pooled data support a relationship between LGE and cardiovascular mortality, heart failure death and all-cause death, but show only a trend towards an increased risk of SCD.144 Late gadolinium enhancement is associated with NSVT on Holter monitoring.140,142
On balance, the extent of LGE on CMR has some utility in predicting cardiovascular mortality, but current data do not support the use of LGE in prediction of SCD risk.
5.5.4 Differential diagnosis
Cardiac magnetic resonance imaging rarely distinguishes the causes of HCM by their magnetic properties alone, but the distribution and severity of interstitial expansion can, in context, suggest specific diagnoses. Anderson-Fabry disease is characterized by a reduction in non-contrast T1 signal and the presence of posterolateral LGE.145,146 In cardiac amyloidosis, there is often global, sub-endocardial or segmental LGE and a highly specific pattern of myocardial and blood-pool gadolinium kinetics caused by similar myocardial and blood T1 signals.22,147 The absence of fibrosis may be helpful in differentiating HCM from physiological adaptation in athletes, but LGE may be absent in people with HCM, particularly the young and those with mild disease.
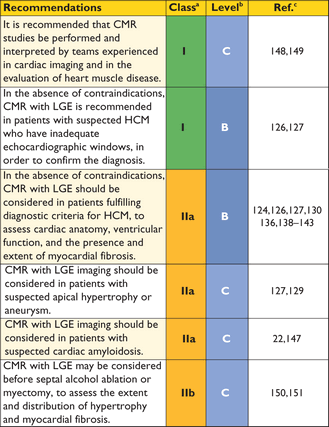
Recommendations for cardiovascular magnetic resonance evaluation in hypertrophic cardiomyopathy
 |
|
CMR = cardiac magnetic resonance; HCM = hypertrophic cardiomyopathy; LGE = late gadolinium enhancement.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for cardiovascular magnetic resonance evaluation in hypertrophic cardiomyopathy
|
|
CMR = cardiac magnetic resonance; HCM = hypertrophic cardiomyopathy; LGE = late gadolinium enhancement.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
5.6 Nuclear imaging and computerized tomography
Nuclear imaging, including positron emission tomography (PET) has been used to measure myocardial blood flow and to detect myocardial perfusion defects in patients with HCM, but its value in the diagnosis of HCM is limited.152–155 The major clinical contribution of nuclear imaging is the detection of TTR-related cardiac amyloidosis. Transthyretin is a tetrameric plasma transport protein synthesized in the liver and is the precursor protein in senile systemic amyloidosis and familial TTR-related amyloidosis.156,157 Several studies have suggested that TTR-derived fibrils show avidity for bone tracers, in particular 99mTechnetium-3,3-diphosphono-1,2-propano-di-carboxylic acid (99mTc-DPD), whereas there is no uptake of tracer in the hearts of patients with HCM caused by sarcomeric protein gene mutations. For this reason, bone scintigraphy (ideally with 99mTc-DPD) should be considered in patients in whom TTR amyloidosis is a possibility (age >65 years, history of bilateral carpal tunnel syndrome, absent family history of HCM, and features consistent with cardiac amyloidosis on ECG and cardiac imaging).156–158
The high contrast resolution of CT provides clear delineation of the myocardium and accurate measurement of wall thickness, ventricular volumes, ejection fraction and LV mass, which correlate well with magnetic resonance imaging, echocardiography and gated SPECT.159 Cardiovascular CT permits the simultaneous imaging of the coronary arteries and valves and can be used to guide catheter ablation of supraventricular arrhythmia.159 Data on myocardial tissue characterization in small cohorts suggest that contrast CT may be useful in the detection of replacement myocardial fibrosis but this requires further study.160,161 Cardiac CT should be considered in patients for whom there are inadequate echocardiographic imaging and contraindications for CMR.159
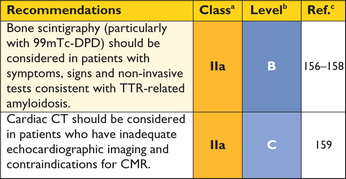
Recommendations for nuclear scintigraphy
 |
|
CT = computerized tomography; 99mTc-DPD = 99mTechnetium-3,3-diphosphono-1,2-propano-di-carboxylic acid; TTR = transthyretin.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for nuclear scintigraphy
|
|
CT = computerized tomography; 99mTc-DPD = 99mTechnetium-3,3-diphosphono-1,2-propano-di-carboxylic acid; TTR = transthyretin.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
5.7 Endomyocardial biopsy
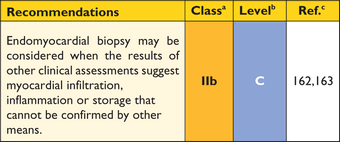
Many of the genetic and non-genetic causes of HCM have characteristic histological appearances, but the diagnosis of HCM is clinical and relies on non-invasive testing in the first instance. As the underlying aetiology can usually be determined using clinical assessment, pedigree analysis, non-invasive imaging, laboratory testing and molecular genetic analysis, endomyocardial biopsy is not part of the routine diagnostic work-up, but it may be considered in clinical scenarios where myocardial infiltration or storage is suspected following specialized tests (including biopsy of other more accessible tissues).162,163
Recommendations for endomyocardial biopsy
 |
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for endomyocardial biopsy
|
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
5.8 Laboratory tests
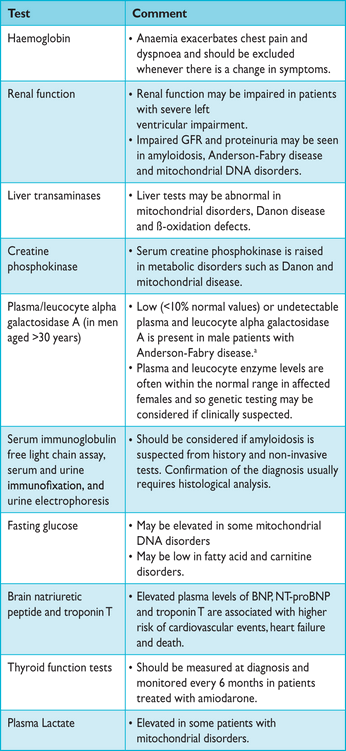
Routine laboratory testing aids the detection of extra-cardiac conditions that cause or exacerbate ventricular dysfunction (for example, thyroid disease, renal dysfunction and diabetes mellitus) and secondary organ dysfunction in patients with severe heart failure. High levels of brain natriuretic peptide (BNP),164 N-terminal pro-brain natriuretic peptide (NT-proBNP)165 and high sensitivity cardiac troponin T (hs-cTnT) are associated with cardiovascular events, heart failure and death. Despite comparable values of ventricular wall thickness, plasma BNP values are three to five times as high in patients with cardiac amyloidosis as those in other causes of HCM. A list of recommended laboratory tests is shown in Table 6.
Recommended laboratory tests in adult patients with hypertrophic cardiomyopathy
 |
|
BNP = brain natriuretic peptide; DNA = deoxyribonucleic acid; GFR = glomerular filtration rate; NT-proBNP = N-terminal pro brain natriuretic peptide.
aPseudo-deficiency may be seen in some genetic variants such as D313Y.166
Recommended laboratory tests in adult patients with hypertrophic cardiomyopathy
|
|
BNP = brain natriuretic peptide; DNA = deoxyribonucleic acid; GFR = glomerular filtration rate; NT-proBNP = N-terminal pro brain natriuretic peptide.
aPseudo-deficiency may be seen in some genetic variants such as D313Y.166
First-line laboratory screening in children is similar to that for adults and should include haematology, glucose, cardiac enzymes (creatine kinase, aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase), renal and liver function tests, pH, electrolytes and uric acid. Following specialist evaluation, additional tests are often required, including measurement of lactate, pyruvate, ammonia, ketones, free fatty acids, carnitine profile, urine organic acids and amino acids.
6. Genetic testing and family screening
In the majority of cases, HCM is inherited as an autosomal dominant genetic trait with a 50% risk of transmission to offspring.34 Some cases are explained by de novo mutations, but apparently sporadic cases can arise because of incomplete penetrance in a parent and, less commonly, autosomal recessive inheritance. In patients fulfilling HCM diagnostic criteria, sequencing of sarcomere protein genes identifies a disease-causing mutation in up to 60% of cases.34,167 The likelihood of finding a causal mutation is highest in patients with familial disease and lowest in older patients and individuals with non-classical features.
6.1 Counselling in probands
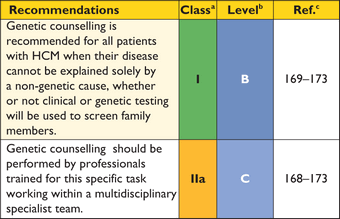
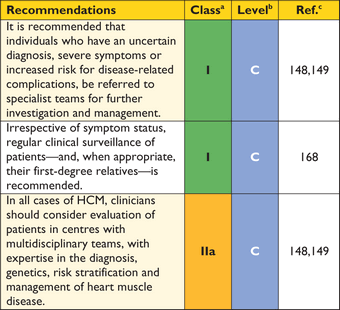
Genetic counselling is recommended in all patients when HCM cannot be explained solely by a non-genetic cause.168
Counselling should be performed by trained healthcare professionals, working within multidisciplinary teams, to help patients understand and manage the psychological, social, professional, ethical and legal implications of a genetic disease.169–173 Counselling also facilitates the gathering of information from other family members, including cardiac and non-cardiac symptoms and autopsy reports that can be used to construct a detailed family pedigree. Pedigree analysis helps to determine the probability of familial disease and the likely mode of inheritance, and provides clues to the underlying aetiology.67 The consequences of a positive test for the patient and their relatives should be explained, and patients should be provided with information on patient support groups and other sources of information including approved websites.
Recommendations on genetic counselling
 |
|
HCM = hypertrophic cardiomyopathy.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on genetic counselling
|
|
HCM = hypertrophic cardiomyopathy.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
6.2 Methods for molecular genetic screening in probands
Conventional genetic practice uses pedigree analysis and clinical evaluation to target molecular testing to the most likely diagnosis. New, high-throughput sequencing (HTS) technologies, capable of analysing entire exomes at similar cost and accuracy to conventional sequencing methods, offer an alternative approach in which no a priori assumptions are made about the cause of disease.174,175 However, screening large numbers of genes results in the identification of many rare non-synonymous genetic variants of unknown significance.175–177 An intermediate approach is the analysis of a pre-defined panel of HCM-related genes using HTS, but the benefit compared with other strategies remains to be determined.19
Irrespective of the sequencing methodology employed, genetic analysis should include the most commonly implicated sarcomere protein genes (Figure 1; Web Table 2). In patients who have features suggestive of specific rare genetic diseases (see section 5) there should be a rational search for pathogenic mutations in other genes. All mutation analyses should comply with the general principles of genetic testing and diagnostic tests should be conducted by certified laboratories using validated methods of genetic analysis and reporting.169–173
6.3 Indications for genetic testing in probands
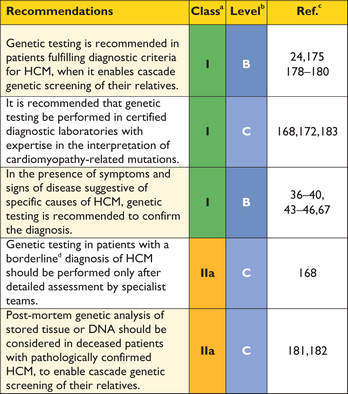
The task force acknowledge that limited resources make implementation of genetic testing challenging in some healthcare systems. Nevertheless, identification of causative mutations facilitates pre-symptomatic diagnosis of family members, clinical surveillance and reproductive advice.
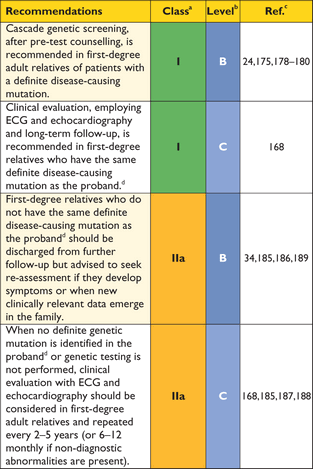
For this reason, genetic testing is recommended in patients fulfilling diagnostic criteria for HCM to enable cascade genetic screening of their relatives.24,175,178–180
The lack of robust data on specific genotype–phenotype associations means that the impact of genetic testing on clinical management is limited mostly to some of the rare genetic causes of HCM. Genetic testing may be of limited clinical value when first-degree relatives are unavailable or unwilling to consider screening for the disease.
Genetic testing in individuals with an equivocal clinical diagnosis (e.g. athletes and hypertensives), should only be performed after detailed clinical and family assessment by teams experienced in the diagnosis and management of cardiomyopathies as the absence of a sarcomere mutation does not exclude familial HCM and variants of uncertain significance are difficult to interpret.168
Genetic analysis of post-mortem tissue or DNA samples can be valuable in the assessment of surviving relatives, but must be interpreted in the light of detailed post-mortem examination of the heart and in accordance with conventional rules for assigning pathogenicity to genetic variants.181,182
Recommendations on genetic testing in probands
 |
|
DNA = deoxyribonucleic acid; HCM = hypertrophic cardiomyopathy.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dBorderline: left ventricular wall thickness 12–13 mm in adults; left ventricular hypertrophy in the presence of hypertension, athletic training, valve disease.
Recommendations on genetic testing in probands
|
|
DNA = deoxyribonucleic acid; HCM = hypertrophic cardiomyopathy.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dBorderline: left ventricular wall thickness 12–13 mm in adults; left ventricular hypertrophy in the presence of hypertension, athletic training, valve disease.
6.4 Genetic and clinical screening of relatives
The legal framework for informing relatives about the presence of a potentially inheritable condition in their family varies considerably around the world. In most countries it is the proband (usually the first person in the family to be diagnosed) and not the clinician, who must inform relatives and invite them for screening on behalf of the healthcare system.184 An information letter is sometimes provided to the patient to help this process.184 Since most relatives have no symptoms at initial clinical screening, it is important that they are provided with information about the consequences of a diagnosis for life insurance, pension, occupation, sporting activities, and eligibility for fostering and adoption before they are tested.
6.4.1 Families with definite disease causing genetic mutations
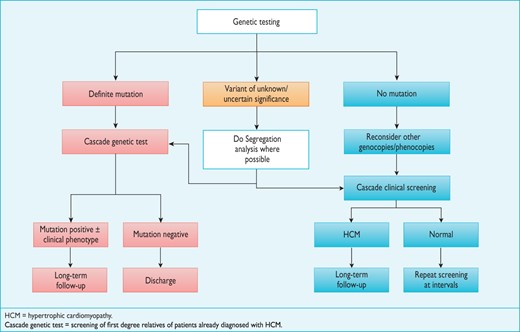
When a definite causative genetic mutation is identified in a patient, his or her relatives should first be genetically tested, and then clinically evaluated if they are found to carry the same mutation(Figure 4).
Flow chart for the genetic and clinical screening of probands and relatives.
Economic decision models have compared the cost-effectiveness of molecular screening to clinical screening alone and have shown that the combination of genetic testing and clinical screening identifies more individuals at risk of developing the disease and allows a greater number to be discharged from follow-up.185,186 For this reason, cascade genetic testing can be offered to all relatives when a definite mutation is identified in the proband. When the mutation is absent, relatives should be discharged from clinic but advised to seek re-assessment if they develop symptoms or if new clinically relevant data emerge in the family. A different approach may be considered in children, to take into account issues of consent and the long-term implications of a positive genetic test. If requested by the parents or legal guardian, clinical evaluation may precede or be substituted for genetic evaluation when this has been agreed to be in the best interests of the child.
6.4.2 Families without definite disease causing genetic mutations
First-degree adult relatives should be offered clinical screening with an ECG and echocardiogram when genetic testing is not performed in the proband, or when genetic analysis fails to identify a definite mutation or reveals one or more genetic variants of unknown significance (Figure 4).168,185,187,188
Importantly, the phenomenon of age-related penetrance means that a normal clinical evaluation does not exclude the possibility of disease development in the future; first-degree relatives should therefore be offered repeat assessment.168
The frequency of clinical screening in the absence of a genetic diagnosis should be guided by the age of onset and severity of cardiomyopathy within the family (e.g. the occurrence of multiple and early sudden deaths) and active participation in competitive sport. Individuals who have non-diagnostic clinical features consistent with early disease should be seen initially at intervals of 6–12 months and then less frequently if there is no progression. All relatives who complain of new cardiovascular symptoms should be re-evaluated promptly.
Recommendations for genetic and clinical testing of adult relatives
 |
|
ECG = electrocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dProband = usually the first family member to be diagnosed with the condition.
Recommendations for genetic and clinical testing of adult relatives
|
|
ECG = electrocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dProband = usually the first family member to be diagnosed with the condition.
6.5 Clinical and genetic screening of children
Clinical and genetic testing of children should be guided by the best interests of each child in accordance with international standards for good practice.190–192 Potential benefits of screening in childhood include reduction of uncertainty and anxiety, psychological adjustment, the opportunity to make realistic life plans, and targeted clinical surveillance. Potential harm includes increased ambiguity if a specific phenotype cannot be predicted, alteration of self-image, distortion of perception of the child by parents and other responsible adults such as teachers, increased anxiety and guilt, and compromised life insurance prospects.
The guiding principle is that a genetic or a clinical test in a child should have an impact on management, lifestyle and further clinical screening.
Prospective clinical data on children with disease causing sarcomere protein gene mutations are limited, but best evidence suggests that clinically important events in asymptomatic children are rare before puberty.189 The consensus view of the committee preparing these Guidelines is that clinical and/or genetic screening should be considered from the age of 10 years onwards. Clinical or genetic testing at a younger age may be appropriate in families with early-onset disorders (for example, disorders of the MAPK pathway, inherited errors of metabolism or multiple sarcomere mutations), when there is a malignant family history in childhood and when children have cardiac symptoms or are involved in particularly demanding physical activity.
Recommendations for genetic and clinical screening in children
 |
|
ECG = electrocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for genetic and clinical screening in children
|
|
ECG = electrocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
6.6 Follow-up of mutation carriers without a phenotype
Preliminary studies suggest that there are no major adverse psychological consequences associated with long-term clinical and genetic screening in children and adults at risk of disease development when they are managed in expert centres.189 There are very few data on the natural histories of individuals who carry a disease-causing mutation and have no phenotype, but recent studies suggest a benign clinical course for most clinically unaffected mutation carriers.189,193 The clinical significance of mild morphological and functional abnormalities is uncertain but probably minor in most cases.194–196 Sudden cardiac death is rare in the absence of cardiac hypertrophy and is confined mostly to isolated reports of patients with troponin T gene mutations.27,28,197,198 Cross-sectional studies suggest age-related increases in penetrance,30,189,199–201 implying that a proportion of clinically unaffected mutation carriers will develop overt cardiomyopathy later in life. Thus, precautionary long-term evaluation of normal healthy mutation carriers is recommended. Mutation carriers without disease expression on ECG or echocardiography, who wish to participate in competitive sports, should be advised on a individual basis, taking into account the local legal framework, the underlying mutation and the type of sporting activity.202
Recommendations for follow-up of mutation carriers without a phenotype
 |
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for follow-up of mutation carriers without a phenotype
|
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
6.7 Pre-implantation and pre-natal genetic testing
(See also section 11.4)
Pre-natal genetic diagnosis can be performed at the beginning of pregnancy using chorionic villus sampling or amniocentesis, but the procedure is not legal in some European countries and is restricted to severe and untreatable diseases in others. Given the considerable variability in the phenotypic expression of HCM and its often benign natural history, pre-natal genetic diagnosis of HCM will rarely be appropriate.168,203 Alternative options to pre-natal diagnosis can be discussed, such as adoption, artificial insemination using donated gametes, and pre-implantation genetic diagnosis.168 Use of foetal echocardiography to detect early disease is not recommended since the probability of cardiac expression in the foetus is extremely low, with the exception of some syndromic and metabolic disorders.
7. Delivery of care
Hypertrophic cardiomyopathy is an 'umbrella' term that encompasses a diverse and complex spectrum of genetic and acquired diseases. As a consequence, the diagnosis and management of patients with HCM requires a range of skills and competencies. In some healthcare systems, a ‘hub and spoke’ model—in which specialist services are concentrated in a small number of central facilities, with less specialist aspects of care provided by district cardiology services—may be the most effective way of providing the necessary range of skills.148,204 In other systems, a less centralized approach may be more practical. Whatever the model used, all patients and families should be managed in accordance with the same internationally agreed standards.
Whilst it is not the remit of this task force to describe in detail systems of care for patients with HCM, adherence to a standard of care is essential if the recommendations of these Guidelines are to be implemented effectively.
Recommendations on delivery of care
 |
|
HCM = hypertrophic cardiomyopathy.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on delivery of care
|
|
HCM = hypertrophic cardiomyopathy.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
7.1 Education and training
With advancing knowledge and greater public awareness of inherited cardiac conditions, the demand for specialist cardiomyopathy services will grow. National societies and healthcare providers should ensure that there is a workforce with the necessary skills to fulfil this need, and provide sufficient educational resources to improve and maintain competencies for all professional groups involved in the care of patients with HCM. National and international societies should also develop registries and care networks for patients with cardiomyopathies.
8. Assessment of symptoms
Most people with HCM are asymptomatic and have a normal lifespan but some develop symptoms, often many years after the appearance of ECG or echocardiographic evidence of LVH. In infants, symptoms and signs of heart failure include tachypnoea, poor feeding, excessive sweating and failure to thrive. Older children, adolescents and adults complain of fatigue and dyspnoea as well as chest pain, palpitations and syncope. Systematic 2D and Doppler echocardiography and ambulatory ECG monitoring are usually sufficient to determine the most likely cause of symptoms. Assessment of LVOTO as outlined in section 5.4 should be part of the routine evaluation of all symptomatic patients.
8.1 Chest pain
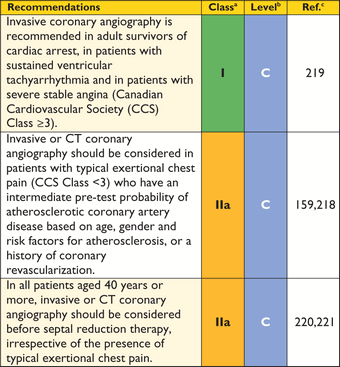
Many patients complain of chest pain at rest or on exertion. Pain may also be precipitated by large meals or alcohol.205–207 The causes of chest pain include myocardial ischaemia due to microvascular dysfunction, increased LV wall stress and LVOTO. Congenital coronary artery anomalies, including tunnelled left anterior descending artery or atherosclerotic coronary artery disease, may also be responsible.208 Systolic compression of epicardial and intramural vessels is very common but is not usually of clinical importance.209–211
Resting ECG abnormalities and a high prevalence of perfusion abnormalities on nuclear imaging and CMR mean that these techniques are of limited use in differentiating obstructive coronary disease from other causes of chest pain and in determining pre-test probability of coronary disease in patients with HCM.212–217 Patients with typical angina on exertion should be considered for invasive or CT coronary angiography on the basis of their symptoms, age, gender and atherosclerosis risk factors, as outlined in existing ESC Guidelines.159,218 Coronary angiography is recommended in adult survivors of cardiac arrest, in patients with sustained ventricular arrhythmia and in symptomatic patients with previous coronary revascularization procedures.219 Invasive or CT coronary angiography should be considered before septal reduction therapy in all patients aged 40 years or more, irrespective of the presence of typical angina.
Recommendations on coronary angiography
 |
|
CT = computed tomography; CCS = Canadian Cardiovascular Society
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on coronary angiography
|
|
CT = computed tomography; CCS = Canadian Cardiovascular Society
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
8.2 Heart failure
Symptoms of chronic heart failure are frequent, but the clinical profile of advanced heart failure varies between patients. In some, heart failure is associated with diastolic dysfunction with preserved EF and small LV size; in others, symptoms are caused by systolic left ventricular dysfunction or LVOTO (with or without mitral insufficiency).222 Atrial fibrillation can complicate any of these scenarios and exacerbate symptoms.223 Recognition of the heterogeneous pathophysiology of heart failure in HCM is important because it influences management.
In most patients, there is a life-long process of progressive and adverse cardiac remodelling, characterized by myocardial fibrosis and wall thinning.222,224,225 In the early stages of this process, patients are often asymptomatic, and conventional non-invasive indices of cardiac performance are within the normal range. As the disease progresses, there is a decline in LV diastolic and systolic function, associated with either mild-to-moderate LV dilation, decreased LV wall thickness, and a fall in LV EF (sometimes referred to as the 'burnt-out' or hypokinetic dilated phase) or severe LV diastolic dysfunction, accompanied by marked atrial dilation with little or no LV dilation (the 'restrictive' phenotype).222 Mitral and tricuspid regurgitation and moderate-to-severe pulmonary hypertension are often present in these advanced stages.226
Presentation with acute heart failure is uncommon, but this can be precipitated by arrhythmias [AF, supraventricular tachycardia (SVT) or sustained ventricular tachycardia (VT)], acute mitral regurgitation (e.g. chordal rupture or infective endocarditis), myocardial ischaemia and infarction, and comorbidity (e.g. anaemia or hyperthyroidism).
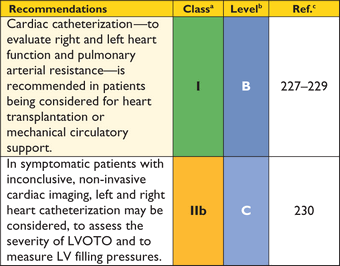
8.2.1 Invasive pressure studies
Non-invasive cardiac imaging has largely replaced cardiac catheterization in the routine assessment of cardiac function. Invasive measurement of intra-cardiac pressures may be appropriate when non-invasive cardiac imaging is insufficient to assess the severity of LVOTO and when planning invasive therapy (e.g. treatment of valve disease) and cardiac transplantation.227
Recommendations on invasive haemodynamic studies
 |
|
LV = left ventricular; LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on invasive haemodynamic studies
|
|
LV = left ventricular; LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
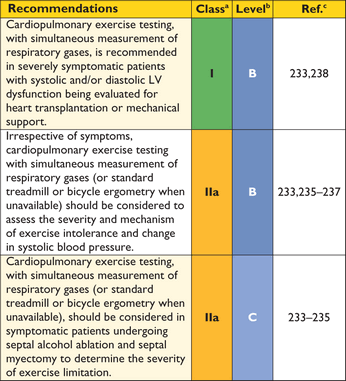
8.2.2 Cardiopulmonary exercise testing
When performed in experienced laboratories, cardiopulmonary exercise testing, with simultaneous measurement of respiratory gases, provides objective information about the severity of functional limitation and its mechanisms. It may be helpful in differentiating HCM from physiological ventricular hypertrophy in athletes and can provide diagnostic clues, such as a disproportionate reduction in peak oxygen consumption and low anaerobic threshold in patients with metabolic disorders.231,232 When facilities are available, cardiopulmonary exercise testing, with simultaneous measurement of respiratory gases, should be considered at the initial clinical evaluation, when patients report a change in symptoms, and when considering invasive LV outflow tract gradient reduction.233–235 Cardiopulmonary exercise testing is recommended in all patients being considered for cardiac transplantation.227
When cardiopulmonary exercise testing is unavailable, conventional treadmill- or bicycle ergometry, with simultaneous electrocardiography, can be used as an alternative. Irrespective of the method of exercise testing, measurement of blood pressure during exercise is recommended, using a standard sphygmomanometer, in order to determine the change in systolic blood pressure that may provide prognostic information (See 9.5: Sudden cardiac death).236,237
Recommendations on cardiopulmonary exercise testing
 |
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
LV = left ventricular
Recommendations on cardiopulmonary exercise testing
|
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
LV = left ventricular
8.3 Syncope
Causes of syncope in HCM include hypovolaemia, complete heart block,239 sinus node dysfunction,239 sustained ventricular tachycardia,LVOTO,240 and abnormal vascular reflexes.237,241,242 Occasionally atrial arrhythmias with fast ventricular response can precipitate syncope, particularly in individuals with preserved atrial function and high filling pressures.223 There can be more than one reason why patients with HCM lose consciousness, including co-morbidities such as epilepsy and diabetes.243
Syncope after prolonged standing in a hot environment, or during the postprandial absorptive state, is suggestive of neurally mediated (reflex) syncope, particularly when it is associated with nausea and vomiting. Syncope during exertion, or immediately following palpitation or chest pain, suggests a cardiac mechanism.243 Provocable obstruction85 should be excluded when patients experience recurrent effort syncope in similar circumstances—for example, when hurrying upstairs or straining. Ventricular arrhythmias are an uncommon cause of syncope, but should be suspected after an unheralded episode, particularly when it occurs at rest or on minimal exertion.
As unexplained non-vasovagal syncope is a risk factor for sudden cardiac death,99,244–,248particularly when it occurs in young patients in close temporal proximity to their first evaluation,99treatment with a prophylactic implantable cardioverter defibrillator (ICD) may be appropriate in individuals with other features indicative of high sudden death risk, even if the mechanism of syncope is undetermined at the end of a complete work-up. The fact that syncope may be caused by mechanisms other than ventricular arrhythmia means that patients may remain at risk of recurrent syncope after ICD implantation.
Patients with syncope should undergo 12-lead ECG, standard upright exercise test and 48-hour ambulatory ECG monitoring and, if a bradyarrhythmia is identified, it should be treated in accordance with current ESC Guidelines on cardiac pacing.249 Exercise stress echocardiography should be considered, particularly in patients with exertional or postural syncope, to detect provocable LVOTO.85 In patients with recurrent episodes of unexplained syncope, who are at low risk of SCD, an implantable loop recorder (ILR) should be considered.249,250 There are few data on tilt testing in patients with HCM, but a high rate of positive tests in patients without a history of syncope suggests that it is not useful in routine assessment unless there are other features to suggest an autonomic mechanism.243,251,252
Recommendations on investigation of syncope
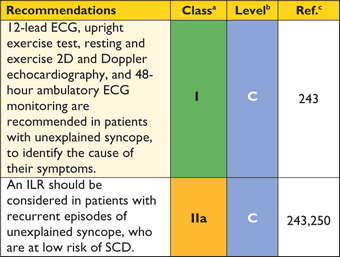 |
|
2D = two-dimensional; ECG = electrocardiogram; ILR = implantable loop recorder; SCD = sudden cardiac death.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on investigation of syncope
|
|
2D = two-dimensional; ECG = electrocardiogram; ILR = implantable loop recorder; SCD = sudden cardiac death.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
8.4 Palpitations
Many patients complain of palpitations,165,246 caused by symptomatic cardiac contractions and ventricular ectopy. A sustained episode of palpitation lasting for more than a few minutes is often caused by supraventricular arrhythmia. In patients with frequent palpitations, 48-hour ambulatory electrocardiography should be performed.250,253 If a cause is not identified, an ILR may be considered.250
Recommendations on palpitations
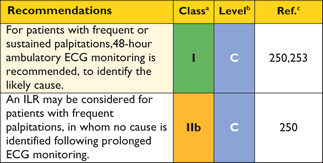 |
|
ECG = electrocardiogram; ILR = implantable loop recorder.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on palpitations
|
|
ECG = electrocardiogram; ILR = implantable loop recorder.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
8.5 Role of electrophysiological testing
The routine use of electrophysiological studies (EPS) in patients with syncope or symptoms suggestive of arrhythmia is not recommended. EPS are indicated in patients with persistent or recurrent supraventricular tachycardia (atrial flutter, atrial tachycardia, atrioventricular nodal re-entry tachycardia, accessory atrioventricular pathway mediated tachycardias) and in patients who have evidence from other non-invasive tests, suggestive of either sino-atrial disease or AV block.249,254 Electrophysiological studies are also indicated when patients have ventricular pre-excitation, to identify and treat an ablatable substrate.255 Invasive EPS may be considered in selected patients with documented, symptomatic, monomorphic, sustained (>30 s) ventricular tachycardia, to identify and treat an ablatable arrhythmia substrate.256,257
Recommendations on electrophysiologic testing
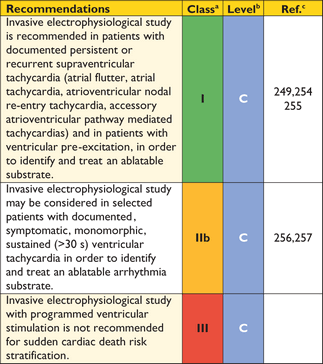 |
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on electrophysiologic testing
|
|
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9. Management of symptoms and complications
In the absence of large randomized trials,2 pharmacological therapy is administered on an empirical basis to improve functional capacity, reduce symptoms and prevent disease progression. In symptomatic patients with LVOTO, the aim is to improve symptoms by using drugs, surgery, alcohol ablation or pacing. Therapy in symptomatic patients without LVOTO focuses on management of arrhythmia, reduction of LV filling pressures, and treatment of angina. Patients with progressive LV systolic or diastolic dysfunction refractory to medical therapy may be candidates for cardiac transplantation.
9.1 Left ventricular outflow tract obstruction
By convention, LVOTO is defined as a peak instantaneous Doppler LV outflow tract gradient of ≥30 mm Hg, but the threshold for invasive treatment is usually considered to be ≥50 mm Hg.
Most patients with a maximum resting or provoked LV outflow tract gradient <50 mm Hg should be managed in accordance with the recommendations for non-obstructive HCM but, in a very small number of selected cases with LV outflow tract gradients between 30 and 50 mm Hg and no other obvious cause of symptoms, invasive gradient reduction may be considered, acknowledging that data covering this group are lacking.
9.1.1 General measures
All patients with LVOTO should avoid dehydration and excess alcohol consumption, and weight loss should be encouraged. Arterial and venous dilators, including nitrates and phosphodiesterase type 5 inhibitors, can exacerbate LVOTO and should be avoided if possible (see also management of hypertension, section 12.2).258 New-onset or poorly controlled AF can exacerbate symptoms caused by LVOTO and should be managed by prompt restoration of sinus rhythm or ventricular rate control.223 Digoxin should be avoided in patients with LVOTO because of its positive inotropic effects.259
Recommendations for treatment of left ventricular outflow tract obstruction: general measures
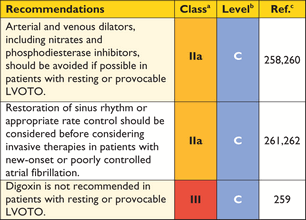 |
|
LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for treatment of left ventricular outflow tract obstruction: general measures
|
|
LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.1.2 Drug therapy
By consensus, patients with symptomatic LVOTO are treated initially with non-vasodilating ß-blockers titrated to maximum tolerated dose, but there are very few studies comparing individual ß-blockers. Small and mostly retrospective studies suggest that oral propranolol can abolish or reduce resting and provocable LVOTO and provide symptomatic benefit263–265 One study has shown improved exercise tolerance and suppression of supraventricular and ventricular arrhythmias in patients treated with sotalol.266
If ß-blockers alone are ineffective, disopyramide (when available), titrated up to a maximum tolerated dose (usually 400–600 mg/day), may be added.267,268 This Class IA anti-arrhythmic drug can abolish basal LV outflow pressure gradients and improve exercise tolerance and functional capacity without proarrhythmic effects or an increased risk of sudden cardiac death.267,268 Dose-limiting anticholinergic side-effects include dry eyes and mouth, urinary hesitancy or retention, and constipation.267,268 The QTc interval should be monitored during dose up-titration and the dose reduced if it exceeds 480 ms. Disopyramide should be avoided in patients with glaucoma, in men with prostatism, and in patients taking other drugs that prolong the QT interval, such as amiodarone and sotalol. Disopyramide may be used in combination with verapamil.268 Disopyramide should be used cautiously in patients with—or prone to—AF in whom drug-induced enhancement of AV conduction can increase the ventricular rate.
Verapamil (starting dose 40 mg three times daily to maximum 480 mg daily) can be used when ß-blockers are contraindicated or ineffective, but close monitoring is required in patients with severe obstruction (≥100 mm Hg) or elevated pulmonary artery systolic pressures, as it can provoke pulmonary oedema.269 Short-term oral administration may increase exercise capacity, improve symptoms and normalize or improve LV diastolic filling without altering systolic function.270–273 Similar findings have been demonstrated for diltiazem (starting dose 60 mg three times daily to maximum 360 mg daily)274 and it should be considered in patients who are intolerant- or have contraindications to ß-blockers and verapamil. Nifedipine and other dihydropyridine calcium antagonists are not recommended for the treatment of LVOTO.275,276
Low-dose loop or thiazide diuretics may be used cautiously to improve dyspnoea associated with LVOTO, but it is important to avoid hypovolaemia.
Beta-blockers should be considered in neonates and children with LVOTO and limited data on verapamil suggest that it can also be used safely in children.272 There are no data on which to make specific recommendations for disopyramide therapy in children. Medical therapy may be considered in asymptomatic or mildly symptomatic adolescents and adults who have a resting or provoked LVOTO and left atrial enlargement.
Rarely, patients with severe provocable LVOTO can present with hypotension and pulmonary oedema that mimics acute myocardial ischaemia. Recognition of this scenario is important, as the use of vasodilators and positive inotropes in this setting can be life-threatening. Treatment should instead consist of oral or i.v. ß-blockers and vasoconstrictors (e.g. phenylephrine, metaraminol and norepinephrine).
Recommendations on medical treatment of LVOTO
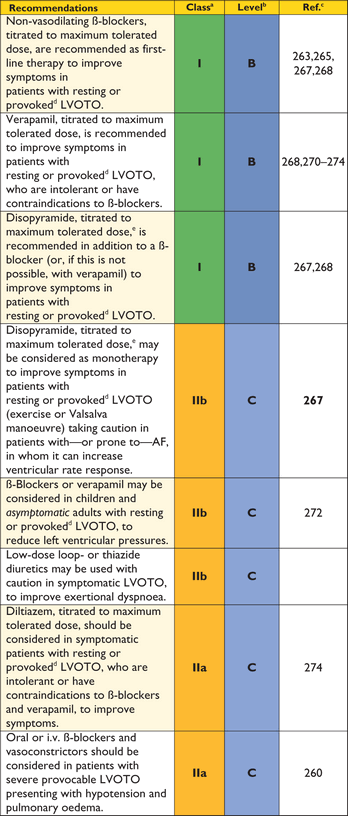 |
|
LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dProvocation with Valsalva manoeuvre, upright exercise or oral nitrates if unable to exercise.
eQTc interval should be monitored during up-titration of disopyramide and the dose reduced if it exceeds 480 ms.
Recommendations on medical treatment of LVOTO
|
|
LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dProvocation with Valsalva manoeuvre, upright exercise or oral nitrates if unable to exercise.
eQTc interval should be monitored during up-titration of disopyramide and the dose reduced if it exceeds 480 ms.
9.1.3 Invasive treatment of left ventricular outflow tract obstruction
There are no data to support the use of invasive procedures to reduce LV outflow obstruction in asymptomatic patients, regardless of its severity.
Invasive treatment to reduce LVOTO should be considered in patients with an LVOTO gradient ≥50 mm Hg, moderate-to-severe symptoms (New York Heart Association (NYHA) functional Class III–IV) and/or recurrent exertional syncope in spite of maximally tolerated drug therapy. In some centres, invasive therapy is also considered in patients with mild symptoms (NYHA Class II) who have a resting or maximum provoked gradient of ≥50 mm Hg (exercise or Valsalva) and moderate-to-severe SAM related mitral regurgitation, AF, or moderate-to-severe left atrial dilation but there are few data supporting this practice.277
9.1.3.1 Surgery
The most commonly performed surgical procedure used to treat LVOTO is ventricular septal myectomy (Morrow procedure), in which a rectangular trough that extends distally to beyond the point of the mitral leaflet–septal contact is created in the basal septum below the aortic valve.278 This abolishes or substantially reduces LV outflow tract gradients in over 90% of cases, reduces SAM-related mitral regurgitation, and improves exercise capacity and symptoms. Long-term symptomatic benefit is achieved in 70–80% of patients with a long-term survival comparable to that of the general population.279–287 Pre-operative determinants of a good long-term outcome are age <50 years, left atrial size <46 mm, absence of atrial fibrillation and male gender.287
The main surgical complications are AV nodal block, ventricular septal defect and aortic regurgitation (AR), but these are uncommon in experienced centres using intraoperative TOE guidance.286,288,289 When there is co-existing mid-cavity obstruction, the standard myectomy can be extended distally into the mid-ventricle around the base of the papillary muscles, but data on the efficacy and long-term outcomes of this approach are limited.290
Concomitant mitral valve surgery is required in 11–20% of patients undergoing myectomy.114 In patients with marked mitral leaflet elongation and/or moderate-to-severe mitral regurgitation, septal myectomy can be combined with one of several adjunctive procedures, including mitral valve replacement, posterior-superior realignment of the papillary muscles, partial excision and mobilization of papillary muscles, anterior mitral leaflet plication, and anterior leaflet extension using a glutaraldehyde-treated pericardial patch that stiffens the mid-portion of the leaflet.291–294 An elongated anterior mitral leaflet favours mitral valve repair instead of replacement.295 Surgical mortality for myectomy with mitral intervention is around 3–4%.294,296,297
9.1.3.2 Septal alcohol ablation
In experienced centres, selective injection of alcohol into a septal perforator artery (or sometimes other branches of the left anterior descending coronary artery) to create a localized septal scar has outcomes similar to surgery in terms of gradient reduction, symptom improvement and exercise capacity.298–302 The main non-fatal complication is AV block in 7–20% of patients and the procedural mortality is similar to isolated myectomy.299–303
Due to the variability of the septal blood supply, myocardial contrast echocardiography is essential prior to alcohol injection. If the contrast agent cannot be localized exclusively to the basal septum at and adjacent to the point of mitral-septal contact, the procedure should be abandoned.111–113
Injection of large volumes of alcohol in multiple septal branches—with the aim of gradient reduction in the catheter laboratory—is not recommended, as it is associated with a high risk of complications and arrhythmic events.304
Alternative methods have been reported in small numbers of patients, including non-alcohol septal embolisation techniques (coils,305,306 polyvinyl alcohol foam particles,307 cyanoacrylate308) and direct endocavitary ablation (radiofrequency, cryotherapy).309,310 These alternative methods have not been directly compared with other septal reduction therapies and long-term outcome/safety data are not available.
9.1.3.3 Surgery vs. alcohol ablation
Experienced multidisciplinary teams should assess all patients before intervention.
The choice of therapy should be based on a systematic assessment of the mitral valve and septal anatomy that includes deliberate exclusion of other LV outflow tract and mitral valve abnormalities requiring surgical treatment. A summary of the key points in pre-operative assessment is shown in Figure 5. Septal ablation may be less effective in patients with extensive septal scarring on CMR and in patients with very severe hypertrophy (≥30 mm), but systematic data are lacking. In general, the risk of ventriculoseptal defect following septal alcohol ablation and septal myectomy is higher in patients with mild hypertrophy (≤16 mm) at the point of the mitral leaflet–septal contact. In such cases, alternatives such as dual chamber pacing (see 9.1.3.5: Dual chamber pacing) or mitral valve repair/replacement may be considered.
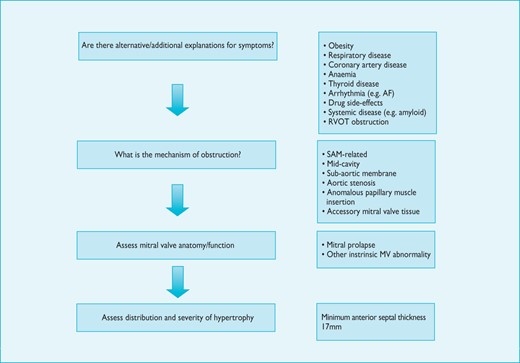
Pre-assessment check list for patients being considered for invasive septal reduction therapies. AF = atrial fibrillation; MV = mitral valve; RVOT = right ventricular outflow tract; SAM = systolic anterior motion.
There are no randomized trials comparing surgery and septal alcohol ablation (SAA), but several meta-analyses have shown that both procedures improve functional status with a similar procedural mortality.311–314 Septal alcohol ablation is associated with a higher risk of AV block, requiring permanent pacemaker implantation and larger residual LV outflow tract gradients.311–314 In contrast to myectomy, most patients develop right-, rather than left bundle branch block after SAA. The risk of AV block following surgery and alcohol ablation is highest in patients with pre-existing conduction disease, and prophylactic permanent pacing before intervention has been advocated.315
The operative mortality of septal myectomy in children is <2% in experienced centres.288 Recurrence of LVOTO requiring re-operation is rare, except in infants and neonates, due to technical limitations of resection and progression of myocardial hypertrophy. Septal alcohol ablation is controversial in children, adolescents and young adults because there are no long-term data on the late effects of a myocardial scar in these groups, and because the technical difficulties and potential hazards of the procedure in smaller children and infants are greater.
Recommendations on septal reduction therapy
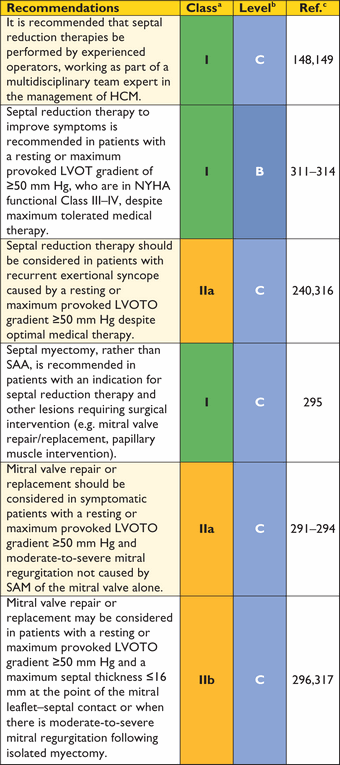 |
|
AF = atrial fibrillation; HCM = hypertrophic cardiomyopathy; LA = left atrium; LVOTO = left ventricular outflow tract obstruction; NYHA = New York Heart Association; SAA = septal alcohol ablation; SAM = systolic anterior motion.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on septal reduction therapy
|
|
AF = atrial fibrillation; HCM = hypertrophic cardiomyopathy; LA = left atrium; LVOTO = left ventricular outflow tract obstruction; NYHA = New York Heart Association; SAA = septal alcohol ablation; SAM = systolic anterior motion.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.1.3.4 Minimum activity requirements
As with other invasive procedures, the results of surgery and alcohol ablation are likely to be better in centres that perform large numbers of procedures. In the absence of specific data, recommendations for the number of interventions per centre and operator are extrapolated from other scenarios. A minimum caseload of 10 SAA and 10 septal myectomies per operator per year is reasonable. More than one trained operator should be available for both procedures, to ensure the safety and sustainability of interventional programmes. National data collection and prospective registries are encouraged to monitor safety and outcomes.
Surgeons and cardiologists who perform invasive gradient reduction therapies should be trained in experienced centres and should work as part of a multidisciplinary team experienced in the management of HCM.
9.1.3.5 Dual chamber pacing
Three small, randomized, placebo-controlled studies of dual chamber pacing and several long-term observational studies have reported reductions in LV outflow tract gradients and variable improvement in symptoms and quality of life.318–322 In one trial, a retrospective subgroup analysis suggested that older patients (>65 years) are more likely to benefit.321 One study has directly compared SAA with pacing and demonstrated superior gradient reduction with ablation.323 A recent Cochrane review concluded that the data on the benefits of pacing are based on physiological measures and lack information on clinically relevant end-points.324
Permanent AV sequential pacing with short AV interval may be considered in symptomatic adult patients who are unsuitable for—or unwilling to consider—other invasive septal reduction therapies, and in patients who have other pacing indications. Pacing parameters should be optimized to achieve maximum pre-excitation of the RV apex with minimal compromise of LV filling (typically achieved with a resting sensed AV interval of 100 ± 30 ms).325 To ensure complete ventricular capture during exercise, a dynamic paced AV interval should be enabled and the programmed upper rate limit should be higher than the fastest sinus rate achieved during exercise.249 Atrioventricular nodal ablation or modification has been advocated as a method for achieving optimal AV programming in some patients with a very short P-R interval, but this is not recommended.326
Recommendations on indications for cardiac pacing in patients with obstruction
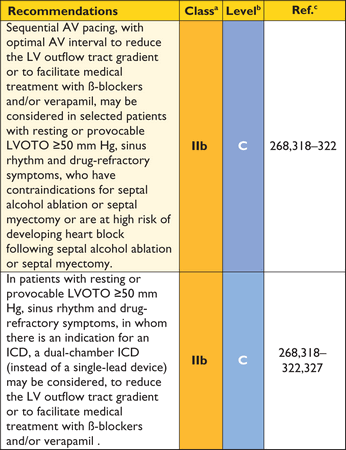 |
|
AV = atrioventricular; ICD = implantable cardioverter defibrillator; LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on indications for cardiac pacing in patients with obstruction
|
|
AV = atrioventricular; ICD = implantable cardioverter defibrillator; LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.2 Left ventricular mid-cavity obstruction and apical aneurysms
LV mid-cavity obstruction occurs in approximately 10% of patients with HCM.328,329 Patients with mid-cavity obstruction tend to be very symptomatic and, in a number of studies, have shown an increased risk of progressive heart failure and SCD.328–330 Approximately 25% of patients also have an LV apical aneurysm which, in some series, is associated with higher cardiovascular mortality.129,328,329,331 Patients with LV mid-cavity obstruction should be treated with high dose ß-blockers, verapamil or diltiazem, but the response is often sub-optimal. Small experience, mostly from single centres, suggests that mid-ventricular obstruction can be relieved by transaortic myectomy, a transapical approach or combined transaortic and transapical incisions, with good short-term outcomes.332,333
LV apical aneurysms by themselves rarely need treatment. A few patients develop monomorphic ventricular tachycardia related to adjacent apical scarring, which may be amenable to mapping and ablation.331,334 Rarely, thrombi are present within the aneurysm and should be treated with long-term oral anticoagulation.335,336 The evidence linking aneurysms to an increased risk of sudden death is confined to small series of selected patients.129 Prophylactic ICD implantation is not recommended in the absence of other clinical features that suggest an increased risk of SCD (see section 9.5).
9.3 Management of symptoms in patients without left ventricular outlow tract obstruction
9.3.1 Heart failure
9.3.1.1 Drug therapy
A general approach to the management of heart failure symptoms is shown in Figure 6. In breathless patients with a normal EF and no evidence of resting or provocable LVOTO, the aim of drug therapy is to reduce LV diastolic pressures and improve LV filling by slowing the heart rate with β-blockers, verapamil or diltiazem (ideally monitored by ambulatory ECG recording) and cautious use of loop diuretics. Restoration of sinus rhythm or ventricular rate control is essential in patients who have permanent or frequent paroxysms of AF (see Atrial tachyarrhythmia, section 9.4) but digoxin is not recommended in patients with preserved EF because of the potentially adverse effects of positive inotropic stimulation.259
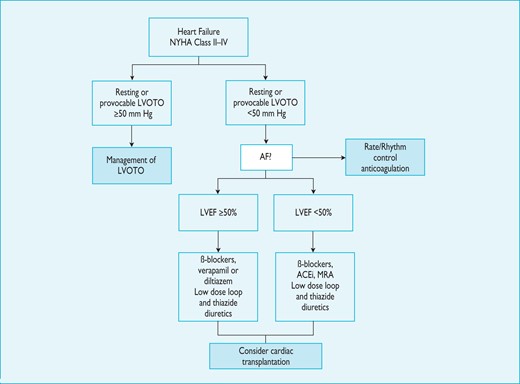
Algorithm for the treatment of heart failure in hypertrophic cardiomyopathy. ACEi = angiotensin converting enzyme inhibitor; AF = atrial fibrillation; LVEF = left ventricular ejection fraction; LVOTO = left ventricular outflow tract obstruction; MRA = mineralocorticoid receptor antagonist; NYHA = New York Heart Association.
Very few studies have examined the effect of renin-angiotensin-aldosterone system (RAAS) inhibition in patients with HCM.2 In the absence of randomized trials, the benefit of RAAS inhibition on hospitalization, symptoms and mortality is assumed, and it is recommended that patients with reduced EF and heart failure symptoms should be treated with diuretics, ß-blockers, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB) and mineralocorticoid receptor antagonists (MRA) in line with the ESC Guidelines for the management of chronic heart failure.337 An EF of <50% is recommended as the threshold for considering therapy with RAAS inhibitors because of the preservation of cavity size in patients with HCM and advanced systolic failure.337 Relatively small LV volumes also mean that some patients may be unable to tolerate high doses of vasodilators and diuretics. In the absence of significant LVOTO, digoxin (0.125 mg–0.5 mg o.d.), alone or in combination with ß-blocker, may be used to control heart rate response in patients with AF and an EF <50%.
Recommendations for patients with heart failure and preserved LV ejection fraction (≥50%)
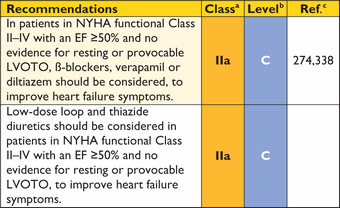 |
|
EF = ejection fraction; LVOTO = left ventricular outflow tract obstruction; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for patients with heart failure and preserved LV ejection fraction (≥50%)
|
|
EF = ejection fraction; LVOTO = left ventricular outflow tract obstruction; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for patients with heart failure and reduced LV ejection fraction (<50%)
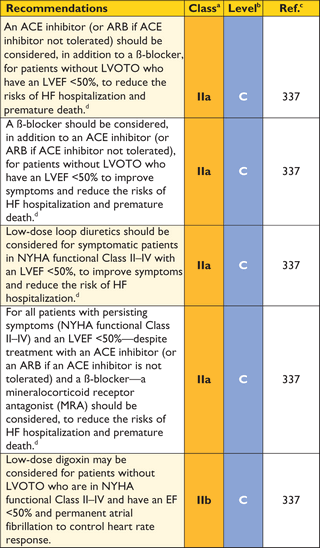 |
|
ACE = angiotensin-converting enzyme; ARB = angiotensin receptor blocker; EF = ejection fraction; HF = heart failure; MRA = mineralocorticoid receptor antagonist; LV = left ventricular; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dIn the absence of randomized trials in HCM, the benefit on hospitalization, symptoms and mortality is assumed but unproven.
Recommendations for patients with heart failure and reduced LV ejection fraction (<50%)
|
|
ACE = angiotensin-converting enzyme; ARB = angiotensin receptor blocker; EF = ejection fraction; HF = heart failure; MRA = mineralocorticoid receptor antagonist; LV = left ventricular; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dIn the absence of randomized trials in HCM, the benefit on hospitalization, symptoms and mortality is assumed but unproven.
9.3.1.2 Cardiac resynchronization therapy
Regional heterogeneity of LV contraction and relaxation is common in patients with HCM and LV dyssynchrony may be a marker of poor prognosis. Case reports and one cohort study have shown that cardiac resynchronization therapy (CRT) can improve heart failure symptoms in patients with left bundle branch block (LBBB) (>120 ms) and is associated with reverse remodelling of the left atrium and LV in patients with impaired LV systolic function.339 In the absence of randomized trials, CRT may be considered in individual patients with refractory symptoms, LV EF <50% and LBBB (QRS duration >120 ms). For patients who have progressed to severe LV dysfunction (EF ≤35%), CRT should be in accordance with current ESC Guidelines.249
Recommendations on cardiac resynchronization therapy
 |
|
EF = ejection fraction; HCM, hypertrophic cardiomyopathy; LBBB = left bundle branch block; LV = left ventricular; LVOTG = left ventricular outflow tract gradient; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on cardiac resynchronization therapy
|
|
EF = ejection fraction; HCM, hypertrophic cardiomyopathy; LBBB = left bundle branch block; LV = left ventricular; LVOTG = left ventricular outflow tract gradient; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.3.1.3 Cardiac transplantation
Orthotopic cardiac transplantation should be considered in patients with moderate-to-severe drug refractory symptoms (NYHA functional Class III–IV) and no LVOTO who meet standard eligibility criteria (see the ESC Guidelines on acute and chronic heart failure).337 HCM accounts for 1–5% of all cardiac transplants performed in the USA and up to 7% of patients on the heart transplantation waiting list in European centres.340 In adolescents and adults, end-stage HCM with LV dilation and systolic dysfunction is the most common clinical profile, with progression to intractable heart failure being more rapid in young patients.341 In infants, massive cardiac hypertrophy with small ventricular cavities and refractory diastolic heart failure is more typical.342 Around 5% of patients referred for cardiac transplantation have refractory ventricular arrhythmia, with or without heart failure symptoms.340 Post-transplant survival is similar to that of other non-HCM indications and superior to that of patients with ischaemic heart disease, with a lower rate of acute rejection.340,341,343,344
Recommendations on cardiac transplantation
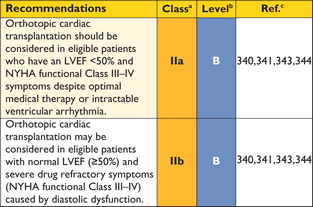 |
|
HF = heart failure; LVEF = left ventricular ejection fraction; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on cardiac transplantation
|
|
HF = heart failure; LVEF = left ventricular ejection fraction; NYHA = New York Heart Association.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.3.1.4 Left ventricular assist devices
As there are increasing numbers of patients with end-stage heart failure and the organ donor pool remains limited, mechanical circulatory support with an LV assist device (LVAD) or biventricular assist device (BiVAD) is increasingly used as a short-term bridge to transplant, or destination therapy in individuals who are not eligible for transplantation. Left ventricular assist devices are rarely used as a bridge to orthotopic heart transplantation in patients with HCM, because their small LV cavities and restrictive LV physiology are thought to preclude device placement.345 However, preliminary data show that patients with HCM and end-stage heart failure may benefit from continuous axial flow LVAD therapy. In one study, right heart failure, prolonged inotropic support, and central venous catheter infections were more common in patients with HCM who were treated with LVADs, but the procedural mortality was comparable to patients with dilated cardiomyopathy and ischaemic heart disease.346 Further research is required in this area but continuous axial flow LVAD therapy may be an option for bridging to therapy in selected patients who are transplant candidates. There are no data on bridge-to-recovery or destination therapy in patients with HCM.
Recommendations on left ventricular assist devices
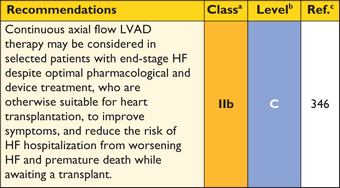 |
|
HF = heart failure; LVAD = left ventricular assist device.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on left ventricular assist devices
|
|
HF = heart failure; LVAD = left ventricular assist device.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.3.2 Angina
ß-Blockers or calcium antagonists should be considered in patients with exertional or prolonged episodes of angina-like pain in the absence of resting or provocable LVOTO or obstructive coronary artery disease. Both classes of drug improve diastolic function and reduce myocardial oxygen demand and, in the case of verapamil, may improve stress-induced sub-endocardial perfusion defects.347–351 In the absence of LVOTO, cautious use of oral nitrates may be considered.
Recommendations for chest pain on exertion in patients without left ventricular outflow tract obstruction
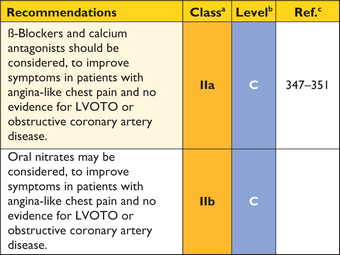 |
|
LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations for chest pain on exertion in patients without left ventricular outflow tract obstruction
|
|
LVOTO = left ventricular outflow tract obstruction.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.4 Atrial tachyarrhythmia
Atrial fibrillation is the most common arrhythmia in patients with HCM. Predisposing factors include increased left atrial pressure and size, caused by diastolic dysfunction, LVOTO and mitral regurgitation. In a recent systematic review, the prevalence and annual incidence of AF were 22.5% and 3.1%, respectively; the prevalence and annual incidence of thromboembolism (stroke and peripheral embolism) in patients with AF were 27.1% and 3.8%.72 Clinical features most closely associated with paroxysmal or permanent AF include age and left atrial enlargement.72 Other possible predictors include LVOTO, P-wave duration >140 ms on signal-averaged ECG, paroxysmal SVT, ST-T changes on baseline electrocardiography, premature ventricular contractions, LGE on CMR, and abnormal coronary flow reserve.72 Reported predictors of thromboembolic events include paroxysmal or chronic AF, severe symptoms (NYHA functional Classes III and IV), older age, increased left atrial volume index, male sex and a history of heart failure admissions.72
As left atrial size is a consistent predictor for AF and stroke in patients with HCM, patients in sinus rhythm with LA diameter ≥45 mm should undergo 6–12 monthly 48-hour ambulatory ECG monitoring to detect AF.
There are fewer data on the prevalence and characteristics of atrial flutter and other atrial arrhythmias but, in general, atrial flutter should be managed conventionally and the risk of thromboembolism considered the same as for AF.
9.4.1 Acute treatment
New-onset AF is frequently associated with heart failure symptoms and so should be treated promptly in accordance with ESC guidelines.261,262 Immediate direct current (DC) cardioversion is recommended in haemodynamically unstable patients.261,262 If patients have severe symptoms of angina or heart failure, intravenous β-blockers or amiodarone are recommended.
In haemodynamically stable patients, oral β-blockers or non-dihydropyridine calcium channel antagonists are recommended to slow the ventricular response to AF.261,262 If pre-excitation is present, non-dihydropyridine calcium channel antagonists and adenosine are contraindicated.261,262 Digoxin should be avoided in patients with LVOTO and normal EF. Similarly, Class IC anti-arrhythmics, such as flecainide and propafenone, should be avoided as they may prolong QRS duration and the QT interval, and increase the ventricular rate due to conversion to atrial flutter and 1:1 ventricular conduction.261,262
When rate control is achieved, elective DC cardioversion should be considered after a minimum of 3 weeks effective [international normalized ratio (INR) between 2.0 and 3.0] anticoagulation with a vitamin K antagonist (VKA). If earlier DC cardioversion is contemplated, a TOE-based strategy should be followed in accordance with ESC guidelines.261,262
9.4.2 Thromboembolism prophylaxis
The ESC Guidelines on stroke prophylaxis in patients with AF recommend a risk factor-based approach in which the risk for patients with non-valvular AF is calculated from a scoring system known as ‘congestive heart failure, hypertension, age ≥75 (doubled), diabetes, stroke (doubled), vascular disease, age 65–74, and female sex' (CHA2DS2-VASc).261,262
As patients with HCM tend to be younger than other high risk groups and have not been included in clinical trials of thromboprophylaxis, use of the CHA2DS2-VASc score to calculate stroke risk is not recommended.
Given the high incidence of stroke in patients with HCM and paroxysmal, persistent or permanent AF, it is recommended that all patients with AF should receive treatment with VKA. In general, lifelong therapy with oral anticoagulants is recommended, even when sinus rhythm is restored.
Two observational studies have reported lower rates of stroke in patients treated with warfarin than in those on antiplatelets or no therapy.223,352 Thus, therapy with a combination of aspirin 75–100 mg and clopidogrel 75 mg daily should be considered only in patients who are unable or unwilling to take oral anticoagulants (OAC). Assessment of the risk of bleeding is recommended when prescribing antithrombotic therapy (whether with VKA, or aspirin in combination with clopidogrel). Even though the ‘hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile INR, elderly (>65 years), drugs/alcohol concomitantly' (HAS-BLED) score was not evaluated in patients with HCM, it would seem to be a reasonable tool to assess the risk of bleeding.353 A HAS-BLED score of ≥3 indicates high bleeding risk and caution should be exercised, with regular clinical reviews.261,262
There are no data on the use of new oral anticoagulants (NOAC) in patients with HCM, but they are recommended when adjusted-dose VKA (INR 2.0–3.0) cannot be used due to a failure to maintain therapeutic anticoagulation or when patients experience side-effects of VKAs or are unable to attend- or undertake INR monitoring. A direct thrombin inhibitor (dabigatran) or an oral factor Xa inhibitor (e.g. rivaroxaban, apixaban) is recommended in this situation. Recommendations for anticoagulation before and after cardioversion are the same as in current ESC guidelines.261,262
9.4.3 Ventricular rate control
Ventricular rate control using β-blockers and non-dihydropyridine calcium channel antagonists—alone or in combination—is recommended in patients with paroxysmal, persistent or permanent AF. 261,262 The choice of medication should be individually determined according to age, lifestyle and heart failure symptoms, and the dose modulated to avoid symptomatic bradycardia but to achieve a resting heart rate <100 BPM. The adequacy of rate control should be assessed during exercise. When adequate rate control cannot be achieved, AV node ablation and permanent pacing may be considered. In the absence of data on the long-term effects of RV pacing on LV function in HCM, the choice of pacing after AV node ablation in patients with persistent or permanent AF should be in line with ESC guidelines, with the exception that CRT-P (CRT with a pacemaker) may be considered in patients with impaired LV function (EF <50%).261,262 In the absence of significant LVOTO, digoxin (0.125 mg–0.5 mg o.d.), alone or in combination with ß-blockers, may be used to control heart rate response in patients with AF and an EF <50%, although data on its efficacy in this context are lacking.
9.4.4 Rhythm control
There are no randomized, controlled trials examining the effect of anti-arrhythmic drugs or radiofrequency ablation on long-term prevention of AF in patients with HCM. One observational study demonstrated that amiodarone therapy was associated with maintenance of sinus rhythm and fewer alterations in drug therapy, embolic episodes and attempted DC cardioversion.354 Others have shown that, in patients variously treated with amiodarone, β-blockers or calcium channel blockers,223,355 there was no significant difference in the duration of sinus rhythm and survival after the first episode of AF. One short-term, double-blind, cross-over study (n =30) demonstrated suppression of supraventricular arrhythmia with sotalol.266 Disopyramide is used to treat LVOTO,267 but its effect on AF suppression in HCM is unknown. There are also no systematic data on the use of dronedarone in patients with HCM but, in view of recent studies showing an increase in cardiovascular events including cardiovascular mortality, it is not recommended in HCM.261,356
There are few data on catheter ablation for AF in patients with HCM,357–361 but the technique should be considered in patients without severe left atrial enlargement, who have drug refractory symptoms or who are unable to take anti-arrhythmic drugs.357 Medium-term maintenance of sinus rhythm is achieved in up to 67% of patients;357–361 failure to suppress AF is associated with left atrial size and older age.357,358
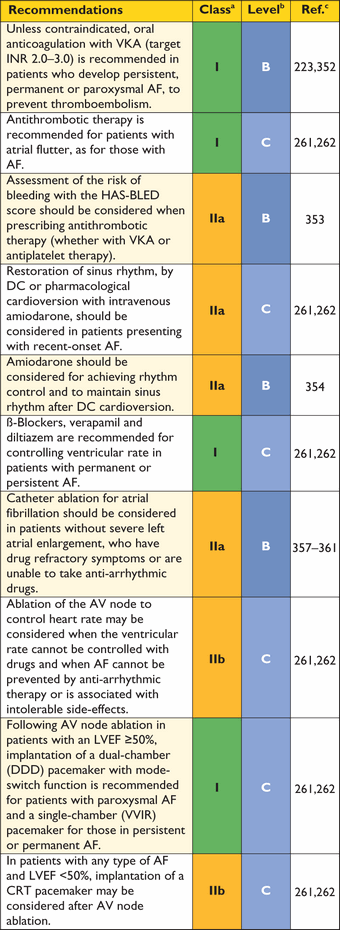
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
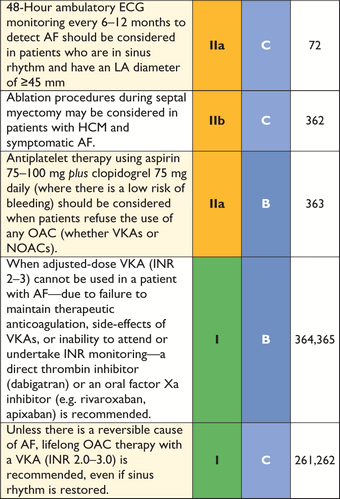 |
|
AF = atrial fibrillation; AV = atrioventricular; CRT = cardiac resynchronization therapy; DC = direct current; ECG = electrocardiogram; HAS-BLED = (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile INR, elderly (>65 years), drugs/alcohol concomitantly); HCM = hypertrophic cardiomyopathy; INR = international normalized ratio; LA = left atrium; NOAC = new oral oral anticoagulant; LVEF = left ventricular ejection fraction; OAC = oral anticoagulant; VKA = vitamin K antagonist;
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
|
|
AF = atrial fibrillation; AV = atrioventricular; CRT = cardiac resynchronization therapy; DC = direct current; ECG = electrocardiogram; HAS-BLED = (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile INR, elderly (>65 years), drugs/alcohol concomitantly); HCM = hypertrophic cardiomyopathy; INR = international normalized ratio; LA = left atrium; NOAC = new oral oral anticoagulant; LVEF = left ventricular ejection fraction; OAC = oral anticoagulant; VKA = vitamin K antagonist;
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.5 Sudden cardiac death
Most contemporary series of adult patients with HCM report an annual incidence for cardiovascular death of 1–2%, with SCD, heart failure and thromboembolism being the main causes of death.366 The most commonly recorded fatal arrhythmic event is spontaneous ventricular fibrillation (VF), but asystole, AV block and pulseless electrical activity are described.239,367–371
9.5.1 Clinical risk assessment
Estimation of SCD risk is an integral part of clinical management. A large body of evidence suggests that, in adolescents and adults, the risk assessment should comprise of clinical and family history, 48-hour ambulatory ECG, TTE (or CMR in the case of poor echo windows) and a symptom-limited exercise test. Clinical features that are associated with an increased SCD risk and that have been used in previous guidelines to estimate risk are shown in Table 7.
Major clinical features associated with an increased risk of sudden cardiac death in adults
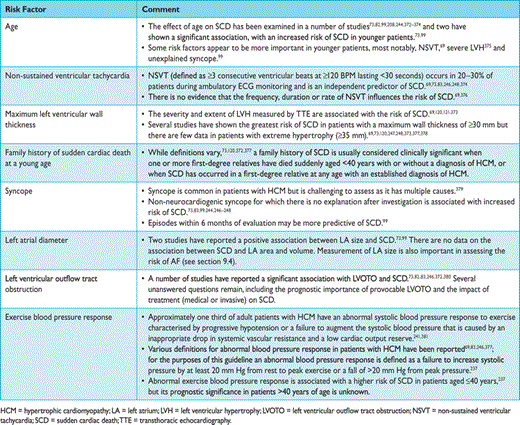 |
|
Major clinical features associated with an increased risk of sudden cardiac death in adults
|
|
9.5.2 Models for estimating sudden cardiac death risk
Trials in other cardiovascular diseases have shown that implantation of an ICD for primary and secondary prophylaxis can reduce mortality;382,383 however, the threshold of risk that justifies device implantation is usually defined by the clinical characteristics of the populations enrolled in such studies, rather than an a priori definition of acceptable risk. This gives rise to a number of inconsistencies as the characteristics of trial populations vary. It is also likely that societal, economic and cultural factors influence the recommendations made by guideline committees.
There are no randomized trials or statistically validated prospective prediction models that can be used to guide ICD implantation in patients with HCM. Recommendations are instead based on observational, retrospective cohort studies that have determined the relationship between clinical characteristics and prognosis.
In the previous version of these Guidelines384 and a more recent guideline from the American College of Cardiology Foundation/American Heart Assocation,385 a small number of clinical characteristics (NSVT, maximal LV wall thickness ≥30mm, family history of SCD, unexplained syncope, and abnormal blood pressure response to exercise) were used to estimate risk and to guide ICD therapy. This approach has a number of limitations: specifically, it estimates relative—and not absolute—risk; it does not account for the different effect size of individual risk factors;386 and some risk factors such as LV wall thickness are treated as binary variables when they are associated with a continuous increase in risk.121 Consequently, current risk algorithms discriminate modestly between high- and low-risk patients.386
Other clinical features, such as myocardial fibrosis (determined by contrast enhanced CMR), LV apical aneurysms and the inheritance of multiple sarcomere protein gene mutations, have been suggested as arbiters that can be used to guide ICD therapy in individuals who are at an intermediate risk, but there are few data to support this approach.33,129,144
Recently, a multicentre, retrospective, longitudinal cohort study of 3675 patients—known as HCM Risk-SCD—developed and validated a new SCD risk prediction model.73 HCM Risk-SCD uses predictor variables that have been associated with an increased risk of sudden death in at least one published multivariable analysis (Web Table 5).73This excludes abnormal blood pressure response as a risk marker. The model provides individualized 5-year risk estimates and, in a head to head comparison with a model using four major risk factors, the performance of the prediction model improved substantially (C-index from 0.54 to 0.7) and compared favourably with other similar prediction algorithms such as CHA2DS2-VASc.73ehu28457
The HCM Risk-SCD formula is as follows:
ProbabilitySCD at 5 years = 1 – 0.998exp(Prognostic index)
wherePrognostic index = [0.15939858 x maximal wall thickness (mm)] − [0.00294271 x maximal wall thickness2 (mm2)] + [0.0259082 x left atrial diameter (mm)] + [0.00446131 x maximal (rest/Valsalva) left ventricular outflow tract gradient (mm Hg)] + [0.4583082 x family history SCD] + [0.82639195 x NSVT] + [0.71650361 x unexplained syncope] − [0.01799934 x age at clinical evaluation (years)].
N.B. In HCM Risk-SCD there was a non-linear relationship between the risk of SCD and maximum left ventricular wall thickness.73This is accounted for in the risk prediction model by the inclusion of a quadratic term for maximum left ventricular wall thickness.
9.5.3 Prevention of sudden cardiac death
9.5.3.1 Exercise restriction
Although documented exercise-induced, sustained, ventricular arrhythmias are rare246—and most ICD therapies for ventricular arrhythmias occur in the absence of tachycardia or physical exertion387,388—patients with HCM should be advised against participation in competitive sports and discouraged from intense physical activity, especially when they have risk factors for SCD and/or LVOTO.
9.5.3.2 Anti-arrhythmic drugs
There are no randomized, controlled data to support the use of anti-arrhythmics for the prevention of SCD in HCM. Amiodarone was associated with a lower incidence of SCD in one small observational study of patients with NSVT on Holter monitoring and in others, increased the threshold for VF, but observational data suggest that amiodarone often fails to prevent SCD.389,390 Disopyramide does not appear to have a significant impact on the risk of SCD.267
9.5.3.3 Implantable cardioverter defibrillators
Secondary prophylaxis
Patients with HCM who survive VF or sustained ventricular tachycardia are at very high risk of subsequent lethal cardiac arrhythmias and should receive an ICD.327,367,391–393 In clinical practice, this population is very small and ICD therapy rarely poses a clinical dilemma.327 There are few data on exercise-induced ventricular arrhythmias but data from one study suggests that it is associated with a high risk of sudden cardiac death.246
Primary prophylaxis
Identification of individuals without a history of VF, who are at high risk of SCD, remains a challenge and only a small subgroup of individuals currently treated with an ICD receives potentially lifesaving shocks.394 At the same time, a large number of ICD recipients experience inappropriate shocks and implant complications.327
In these Guidelines, it is recommended that patients undergo a standardized clinical evaluation (seeWeb Table 5 and Figure 7) that records a pre-defined set of prognostic variables, which are then used to estimate the 5-year risk of SCD using the HCM Risk-SCD model [a Web-based calculator is provided with these Guidelines (www.escardio.org/guidelines-surveys/esc-guidelines/Pages/hypertrophic-cardiomyopathy.aspx)]. The published HCM Risk-SCD dataset has been used to construct three categories of risk (high, intermediate and low) that were determined by consensus (Figure 7). The recommendations for ICD therapy in each risk category take into account not only the absolute statistical risk, but also the age and general health of the patient, socio-economic factors and the psychological impact of therapy. The recommendations are meant to be sufficiently flexible to account for scenarios that are not encompassed by the HCM Risk-SCD model.
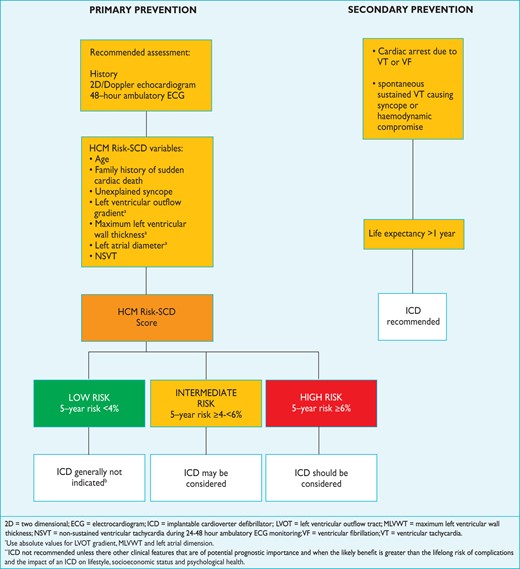
{kind=link}
Flow chart for ICD implantation.
Recommendations on prevention of sudden cardiac death
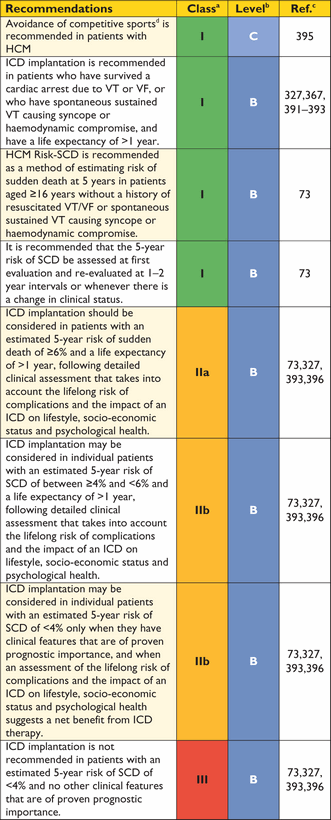 |
|
ICD = implantable cardioverter defibrillator; VF = ventricular fibrillation; VT = ventricular tachycardia.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dESC Guidelines define competitive sport as amateur or professional engagement in exercise training on a regular basis and participation in official competitions (see relevant ESC Guidelines for more detail).395
Recommendations on prevention of sudden cardiac death
|
|
ICD = implantable cardioverter defibrillator; VF = ventricular fibrillation; VT = ventricular tachycardia.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dESC Guidelines define competitive sport as amateur or professional engagement in exercise training on a regular basis and participation in official competitions (see relevant ESC Guidelines for more detail).395
HCM Risk-SCD should not be used in patients <16 years of age, elite athletes or in individuals with metabolic/infiltrative diseases (e.g. Anderson-Fabry disease) and syndromes (e.g. Noonan syndrome). The model does not use exercise-induced LV outflow tract gradients and has not been validated before and after myectomy or alcohol septal ablation.
As the relationship between maximum LV wall thickness and risk is non-linear, the calculated risk for SCD falls in patients with severe LVH (≥35 mm). This may reflect small numbers in this category but, as shown in the source paper, the rate of sudden deaths was very low in this group. This phenomenon has been seen in at least one previous study.99
Pending further studies, HCM-RISK should be used cautiously in patients with a maximum left ventricular wall thickness ≥35 mm.
Implantable cardioverter defibrillators are not recommended when the estimated 5-year risk of SCD is <4% and there are no other clinical features that are of potential prognostic importance (for example multiple young sudden deaths in a family or an abnormal exercise blood pressure response). When such features are present, decisions on ICDs must be made on an individual basis and must balance the likely benefit against the lifelong risk of complications and the impact of an ICD on lifestyle, socio-economic status and psychological health.
Practical aspects of ICD therapy
Prior to ICD implantation, patients should be counselled on the risk of inappropriate shocks, implant complications, and the social and occupational implications (including driving restrictions) of an ICD. Studies examining the role of defibrillation testing at the time of implantation are continuing, but high defibrillation thresholds are reported in patients with severe LVH and in those on amiodarone treatment. 397–400 Until data specific to HCM are available, defibrillation testing may be considered at the physician's discretion. For patients who have a high defibrillation threshold or who fail to cardiovert on defibrillation testing, options include sub-pectoral implantation and standard manoeuvres such as reversal of shocking vector polarity, changing the shock tilt, including/excluding a superior vena cava coil followed by re-testing and, if necessary, implantation of a subcutaneous array.
The VF zone of the device should be programmed at >220/min to minimize shocks from rapidly conducted AF. An SVT discrimination zone, tailored to individual patient characteristics, may also be considered. Observational data show that anti-tachycardia pacing is effective in terminating ventricular arrhythmias in HCM but does not reduce the incidence of appropriate shocks.387,401 As atrial leads do not reduce the incidence of inappropriate shocks,327,393,396 most patients require only a single ventricular lead. Exceptions include patients with LVOTO, in whom an atrial lead provides the option for a short AV delay pacing, and patients in sinus rhythm with impaired LV systolic function, in whom CRT might be preferable (see section 9.3.1.2). In the light of results from the Multicenter Automatic Defibrillator Implantation Trial—Reduce Inappropriate Therapy (MADIT-RIT), shock-only programming can be considered in primary prevention, although this trial was conducted in patients with low EF.402
ß-Blockers and/or amiodarone are recommended in patients with an ICD, who continue to have symptomatic ventricular arrhythmias or recurrent shocks despite optimal treatment and device re-programming.219 Electrophysical study is recommended in patients with ICDs and inappropriate shocks due to regular supraventricular tachycardias, in order to identify and treat any ablatable arrhythmia substrate.403
The newly developed subcutaneous ICD lead system (S-ICD™, Boston Scientific) has FDA approval and may be considered in HCM patients who have no indication for pacing.404 Particular attention should be paid to ensuring optimal R-wave sensing at rest and on exercise, in order to avoid inappropriate shocks from T-wave oversensing. Each patient should have more than one ECG vector that passes screening, to allow alternative programming if oversensing does occur.405,406 Data from a multicentre registry that included 58 HCM patients provided preliminary efficacy and safety data.407
Recommendations on practical aspects of implantable cardioverter defibrillator therapy
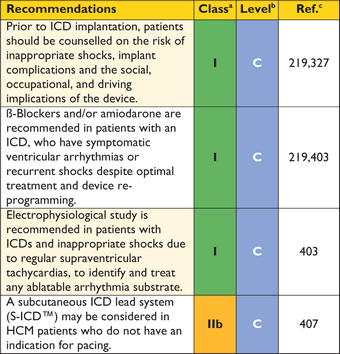 |
|
HCM = hypertrophic cardiomyopathy; ICD = implantable cardioverter defibrillator; S-ICD™= subcutaneous ICD lead system
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on practical aspects of implantable cardioverter defibrillator therapy
|
|
HCM = hypertrophic cardiomyopathy; ICD = implantable cardioverter defibrillator; S-ICD™= subcutaneous ICD lead system
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
9.5.4 Risk of sudden death in children
Implantation of an ICD (epicardial if necessary) is indicated after a life-threatening ventricular arrhythmia in children. In very young children (<8 years old), clinical risk stratification to determine the need for primary prophylaxis with an ICD is hampered by a lack of data. The risk of death or heart transplantation is greatest in infants or in patients with inherited metabolic disorders and malformation syndromes.408 There is general agreement that, as in adults, severe LVH, unexplained syncope, NSVT and a family history of sudden death represent major risk factors for sudden cardiac death.409 The definition of severe hypertrophy in infants, children and pre-adolescents has been assessed using different approaches and measurements.410,411The consensus view for these Guidelines is that a maximum left ventricular wall thickness ≥30 mm or a Z-score ≥6 is considered to be a major risk factor in children.410
Implantation of an ICD should be considered in children who have two or more major risk factors. Single-chamber defibrillators suffice in the majority of cases and reduce the likelihood of complications.412 In individual patients with a single risk factor, ICD implantation may be considered after careful consideration of the risks and benefits to the child and family.
Recommendations on implantation of cardioverter defibrillators in children
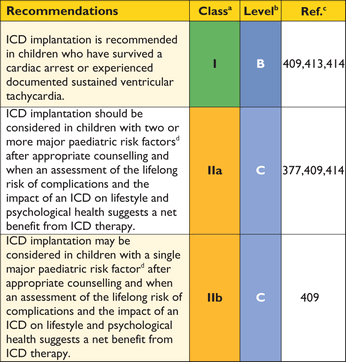 |
|
ICD = implantable cardioverter defibrillator; HCM = hypertrophic cardiomyopathy; SCD = sudden cardiac death.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dMajor paediatric risk factors: Maximum left ventricular wall thickness ≥30 mm or a Z-score ≥6, unexplained syncope, non-sustained ventricular tachycardia (≥3 consecutive ventricular beats at ≥120 BPM lasting <30 seconds), family history of SCD (one or more first-degree relatives with SCD aged <40 years with or without the diagnosis of HCM, or SCD in a first-degree relative at any age with an established diagnosis of HCM).
Recommendations on implantation of cardioverter defibrillators in children
|
|
ICD = implantable cardioverter defibrillator; HCM = hypertrophic cardiomyopathy; SCD = sudden cardiac death.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dMajor paediatric risk factors: Maximum left ventricular wall thickness ≥30 mm or a Z-score ≥6, unexplained syncope, non-sustained ventricular tachycardia (≥3 consecutive ventricular beats at ≥120 BPM lasting <30 seconds), family history of SCD (one or more first-degree relatives with SCD aged <40 years with or without the diagnosis of HCM, or SCD in a first-degree relative at any age with an established diagnosis of HCM).
9.6 Symptomatic bradycardia and atrioventricular block
Symptomatic bradycardia caused by sinus node dysfunction and AV block is relatively uncommon in HCM and should be treated in accordance with the current ESC Guidelines.249 The presence of AV block should raise suspicion of particular genetic subtypes (desmin, FHL1, PRKAG2) in younger patients or amyloidosis and Anderson-Fabry disease in older patients (see section 5 on diagnosis). In contrast, chronotropic incompetence is quite common (particularly in Anderson-Fabry disease) and is an important cause of exercise limitation.415 If AV block is caused by AV node blocking drugs, their dose should be adjusted and the need for pacing re-evaluated.
The benefit of rate-responsive pacing in treating exercise intolerance is uncertain. The risks of chronic RV pacing in HCM with respect to LV systolic function are unknown, but ventricular pacing should be minimized where possible unless treating LVOTO. CRT-P should be considered in patients with impaired systolic function (EF <50%).339,416
9.7 Ventricular tachycardia
Non-sustained ventricular tachycardia (defined as three or more ventricular extrasystoles at a rate of ≥120 BPM, lasting <30 seconds) is a common finding on ambulatory ECG monitoring.69,70,417,418 Its prevalence increases with age and correlates with LV wall thickness and the presence of late gadolinium enhancement on CMR.69,140 Non-sustained ventricular tachycardia is a risk factor for SCD, but does not usually require anti-arrhythmic therapy. Its occurrence during or immediately following exercise is very rare, but may be associated with a high risk of SCD.246
Documented sustained monomorphic VT (≥30 seconds) is uncommon but may be more frequent in patients with apical LV aneurysms.256,419 Exclusion of coronary artery disease should be considered in patients with prolonged or symptomatic episodes and risk factors for coronary atherosclerosis. There is no evidence that haemodynamically tolerated, sustained VT carries a worse prognosis than NSVT but it should be considered as a risk factor for SCD. Patients with poorly tolerated VT should be considered for ICD therapy and treatment with ß-blockers or amiodarone, to suppress further episodes. In patients with evidence for a focal origin, EPS and ablation may be considered.420–422
10. Recommendations for routine follow-up
In general, patients with HCM require lifelong follow-up to detect changes in symptoms, risk of adverse events, LVOTO, LV function and cardiac rhythm.
There are very few longitudinal data on the rates of change in symptoms or cardiac function, but cross-sectional studies show that the prevalence of LV systolic dysfunction and atrial arrhythmia increases with advancing age.222,224,225,423 The frequency of monitoring is determined by the severity of disease, age and symptoms. A clinical examination, including 12-lead ECG and TTE, should be performed every 1–2 years, or sooner should patients complain of new heart failure symptoms. Ambulatory electrocardiography is recommended every year (or every 6 months in the presence of left atrial dilation ≥45 mm) to detect asymptomatic atrial and ventricular arrhythmia, and is indicated whenever patients experience syncope or palpitations.
When available, cardiopulmonary exercise testing can provide objective evidence for worsening disease but need only be performed every 2–3 years unless there is a change in symptoms. There are few data on changes in myocardial fibrosis on CMR during follow-up but, when available, CMR evaluation may be considered every five years or every 2–3 years in patients with progressive disease.424
A complete assessment, including ECG, TTE and ambulatory ECG monitoring should be performed within 1–3 months and at 6–12 months following invasive septal reduction therapies.
Recommendations on routine follow-up
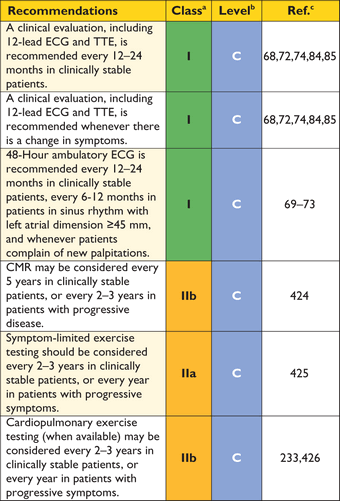 |
|
CMR = cardiac magnetic resonance; ECG = electrocardiogram; TTE = transthoracic echocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
Recommendations on routine follow-up
|
|
CMR = cardiac magnetic resonance; ECG = electrocardiogram; TTE = transthoracic echocardiogram.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
11. Reproduction and contraception
11.1 Introduction
Pregnancy is associated with many physiological changes, including a 40–50% increase in plasma volume and cardiac output, a reduction in systemic vascular resistance and a hypercoagulable state. Whilst most women with HCM have uncomplicated pregnancies, these physiological changes are associated with increased risk for mother and foetus, as the volume load can be poorly tolerated in the setting of LVOTO and diastolic dysfunction. Adequate and timely counselling on contraception, the risks associated with pregnancy, and the risk of disease transmission to the foetus is important in all women with HCM.427,428
11.2 Contraception and termination of pregnancy
When a girl with HCM reaches fertility, she should be counselled to use safe and effective contraception, since unplanned pregnancy carries an increased risk.427 Barrier methods (condoms, diaphragms, pelvic ring) are safe but not as effective as oral contraceptives, even when combined with spermicides (failure rate at 1 year 15–30%). Low dose oral contraceptives with 20 µg or 30 µg ethinyl-oestradiol are effective (though less so in adolescents) and can be safely used in most women with HCM, except for those with increased thromboembolic risk (e.g. women with heart failure or AF), unless they are using adequate oral anticoagulation therapy. Oral contraceptives are inadvisable in any women who smoke and are older than 35 years or who have a history of venous thromboembolism. Emergency contraception is safe for women with HCM.427,429 Progesterone-only contraceptives are a safe alternative but the effectiveness of the progesterone-only pill (desogestrel) depends on compliance (daily intake with <12 hours of variation). Other progesterone-only contraceptives can be used, including 3-monthly injections with medroxyprogesterone acetate or dermal progesterone implants, but should be used cautiously in women with diastolic or systolic heart failure because of the risk of fluid retention. The levonorgestrel-releasing intra-uterine device (IUD) is a safe and effective alternative. Progesterone-only methods are not always tolerated because of irregular blood loss. A copper IUD may be used but is less effective and is associated with an increase in monthly blood loss. Antibiotic prophylaxis at the time of IUD insertion is not necessary, but vasovagal reactions can occur during implantation and, in women with severe LVOTO, it should therefore be implanted after cardiology consultation and in a hospital setting.
Sterilization can be safely accomplished by tubal ligation, although the risks of anaesthesia and abdominal inflation should be considered. Hysteroscopic sterilization with an Essure™ device (Bayer) is an alternative but can also be associated with vasovagal reactions.427,429
Termination of pregnancy should be performed in-hospital after consultation with a cardiologist. Dilation and evacuation are usually safe; as prostaglandin E1 or E2 can lower systemic vascular resistance and increase heart rate, haemodynamic monitoring is indicated. Prostaglandin F increases pulmonary artery pressure and should be avoided.427
11.3 Infertility treatment
In vitro fertilization can be associated with fluid retention and with arterial and venous thromboembolism. It is probably safe in low-risk HCM patients, but should be avoided in patients with heart failure or AF and in women with severe hypertrophy and restrictive LV filling pattern.427 When pre-implantation genetic diagnosis (see section 6) is an issue, the risk of in vitro fertilization should be taken into account.
11. 4 Pre-conception counselling
Most women with HCM tolerate pregnancy well. The hypertrophied small left ventricle can, in most cases, accommodate the physiological increase in blood volume without undue rise in filling pressures. The few reported cases of maternal death have occurred mostly in women who were known to be at very high risk.430–432 Deterioration during pregnancy most often occurs in women who are symptomatic before pregnancy.431,433,434 The prevalence of heart failure during pregnancy differs between studies but it is probably more likely in women who had impaired LV function before pregnancy.431–433,435 Left ventricular outflow tract gradients tend to increase slightly during pregnancy and high pre-pregnancy LVOT gradients have been reported to be associated with more pregnancy complications.430–432,434,436 Women with arrhythmias pre-pregnancy are more likely to experience recurrence in pregnancy,430,436 but pregnancy per se does not seem to substantially increase the risk of arrhythmia.430,432,433,436
Ideally, risk assessment should be performed before conception, using the modified World Health Organization (WHO) classification.427 Most HCM patients are WHO Class II or III (Table 8).427 Justification for advising against pregnancy (WHO Class IV) is present in a small minority with significant LV dysfunction or severe symptomatic LVOTO. Pregnancy may be possible after relief of LVOTO.
Echocardiography should be performed to evaluate ventricular function, mitral regurgitation and LVOTO. Exercise testing (preferably pre-pregnancy or, in asymptomatic pregnant women, to 80% of predicted maximal heart rate) is an important tool to assess functional capacity, heart rate response and arrhythmias.427,437 A plan for use of medication and follow-up during pregnancy should be made and discussed with the patient and her partner before conception.427 Genetic counselling is recommended in all women with HCM (see section 6).
11.5 Management of pregnancy and delivery
Women in WHO Class II should be assessed each trimester. Women in WHO Class III should be followed monthly or bimonthly, in specialized centres, by a multidisciplinary team.427 The focus should be on symptomatic status, LV outflow obstruction, arrhythmias and ventricular function. Echocardiography should be performed each trimester or when new symptoms occur.
Recommendations for drug use during pregnancy and breastfeeding are summarized in Web Table 6.427 When medication is prescribed, possible harmful effects to the foetus need to be considered. However, both doctor and patient should realise that withholding medication from the mother may seriously threaten her health and therefore also the foetus (e.g. treatment of serious ventricular arrhythmias and anticoagulation therapy for AF). Ultimately, the interests of the mother should prevail.
β-Blockers should be continued if already used before pregnancy (though re-evaluation of the need for their use is recommended) and side-effects such as growth retardation, neonatal bradycardia or hypoglycaemia are usually not severe and can be easily managed. β-Blockers should be started when new symptoms occur.427,434 Metoprolol is the most widely used; atenolol is not advised because it has been associated with more growth retardation. Whenever ß-blockers are prescribed, monitoring of foetal growth and of the condition of the neonate is recommended.
Verapamil and diltiazem are classified by the FDA as class C, meaning that their potential benefits may warrant their use in pregnant women despite potential risks.
Disopyramide should only be used when potential benefits outweigh risks, since it can cause uterine contractions.438
Amiodarone should only be used when absolutely necessary, because of risk of the foetal thyroid toxicity, growth retardation, and neurological adverse effects.427,439,440
Poorly tolerated AF can be safely cardioverted during pregnancy. Since a few cases of foetal distress immediately following electrical cardioversion have been described, this procedure should be carried out with facilities available for cardiac monitoring and emergency caesarean section.441 Therapeutic anticoagulation with low molecular weight heparin with anti-factor-Xa monitoring (peak anti-Xa level 0.8–1.2 U/mL 4–6 hours post-dose) in the first trimester and from the 36th week onwards—or VKAs in the second and third trimester—is recommended for paroxysmal or persistent AF.427 New oral anticoagulants (e.g. dabigatran, rivaroxaban) are not advised because of proven toxicity in animals and insufficient data in humans. When indicated, pacemaker or ICD implantation during pregnancy should be performed, if possible with echocardiographic guidance.
A delivery plan should be made at the end of the second trimester by the multidisciplinary team. Planned vaginal delivery is generally preferred although asymptomatic women with mild disease may go into spontaneous labour. Caesarean section is mainly performed for obstetric indications but should be considered in patients with severe LVOTO, pre-term labour while on OAC, or severe heart failure. Epidural and spinal anaesthesia are beneficial for reduction of pain and stress but must be applied judiciously to avoid vasodilation and hypotension, especially when LVOTO is severe. Single-shot spinal anaesthesia should be avoided.427,442 During delivery, monitoring of heart rate and rhythm should be considered in patients with a high risk of developing arrhythmias.434 Oxytocin should only be given as a slow infusion, to avoid hypotension and tachycardia. Because of increased risk of pulmonary oedema due to fluid shifts post-delivery, clinical observation should be continued for 24–48 hours.427 There is no need to deactivate an ICD during vaginal delivery.
Modified WHO classification of maternal cardiovascular risk: principles and application
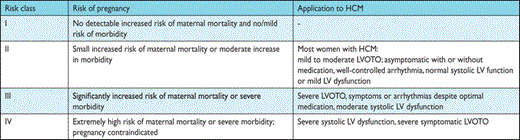 |
|
HCM = hypertrophic cardiomyopathy; LV = left ventricle; LVOTO = left ventricular outflow tract obstruction; WHO = World Health Organization.
Modified WHO classification of maternal cardiovascular risk: principles and application
|
|
HCM = hypertrophic cardiomyopathy; LV = left ventricle; LVOTO = left ventricular outflow tract obstruction; WHO = World Health Organization.
Recommendations on reproductive issues in women with hypertrophic cardiomyopathy
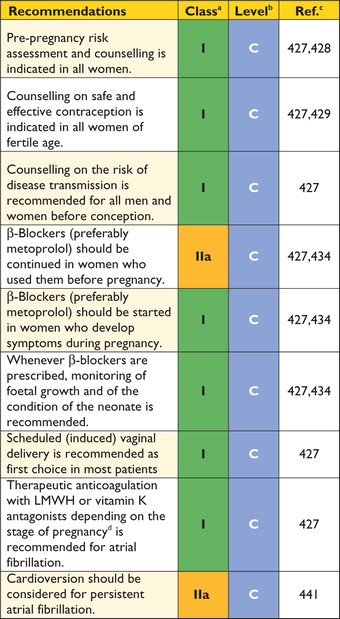 |
|
LMWH = low molecular weight heparin.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dSee text for details
Recommendations on reproductive issues in women with hypertrophic cardiomyopathy
|
|
LMWH = low molecular weight heparin.
aClass of recommendation.
bLevel of evidence.
cReference(s) supporting recommendations.
dSee text for details
12. Special issues
12.1 Diagnosis of hypertrophic cardiomyopathy in athletes
Physiological adaptation to regular intense physical training is associated with ECG manifestations that reflect increased vagal tone, enlarged cardiac chamber size and an increase in LV wall thickness and mass.443 The ability to reliably differentiate between HCM and this normal training effect is important, as an incorrect diagnosis has far-reaching implications for individual athletes and their families, as well as for sporting organizations and society as a whole. Several clinical features that distinguish physiological from pathological hypertrophy have been described,444,445 but dilemmas can arise in individuals with borderline or mild LVH.446 In the absence of a validated ‘gold standard’, the diagnosis of HCM in an athlete requires integration of a number of different parameters of varying sensitivity and specificity. Web Table 7 summarizes the features that assist in differenting between HCM and physiological LV hypertrophy caused by physical training, and that are best supported by published data.59,445–460
12.2 Hypertension
In clinical practice, it can be a challenge to make a differential diagnosis between hypertensive heart disease on the one hand and HCM associated with systemic hypertension on the other. Regression of LVH with treatment of hypertension argues against the diagnosis of HCM, but the reverse is not necessarily true.461–466 Clinical features that suggest a diagnosis of HCM in a patient with hypertension are summarized in Table 9.
12.2.1 Imaging
Increased LV mass, determined by echocardiography, is present in >30% of hypertensive patients.467 The degree of hypertrophy is influenced by ethnicity, neurohumoral factors, and genetic variants.468–470 In general, maximal LV wall thickness is greater in patients with unequivocal HCM, but there is overlap between the two conditions.471–473 The majority of patients with hypertensive LVH have a maximal interventricular septal thickness <15mm,474–477 but in black patients (particularly in the presence of chronic kidney disease) maximal interventricular septal thickness is not uncommonly between 15 and 20 mm.478 Late gadolinium enhancement is reported in the mid-myocardium and epicardium in both hypertension and HCM,136 but tends to be located in the segment with the greatest wall thickness and at the RV insertion points in HCM.136 Similarly, while diastolic abnormalities and LA dilation are seen in HCM and hypertension, severe diastolic dysfunction is more typical of HCM. Doppler myocardial imaging and strain imaging may help to distinguish the two entities.479,480 Resting or exercise-induced LVOTO can be observed in hypertension and does not constitute a diagnostic criterion.481,482
12.2.2 Electrocardiogram
On the 12-lead ECG, LVH by voltage criteria is seen in 10–20% of hypertensive patients with LVH, but—at least in Caucasians—marked repolarisation abnormalities, conduction abnormalities and Q-waves are unusual.68,467,483,484 Atrial fibrillation is common in both conditions, affecting approximately one-third of patients. Premature ventricular complexes and NSVT are reported in up to 30% of patients with hypertension complicated by LVH.485–487
Clinical features that assist in the differential diagnosis of hypertensive heart disease and hypertrophic cardiomyopathy
 |
|
ECG = electrocardiogram; CMR = cardiac magnetic resonance imaging; HCM = hypertrophic cardiomyopathy; LV = left ventricle; LVH = left ventricular hypertrophy; RV = right ventricle.
Clinical features that assist in the differential diagnosis of hypertensive heart disease and hypertrophic cardiomyopathy
|
|
ECG = electrocardiogram; CMR = cardiac magnetic resonance imaging; HCM = hypertrophic cardiomyopathy; LV = left ventricle; LVH = left ventricular hypertrophy; RV = right ventricle.
12.3 Isolated basal septal hypertrophy (sigmoid septum) in elderly people
Some elderly people have mild basal septal hypertrophy (sometimes referred to as a sigmoid septum or septal bulge) associated with increased angulation between the aorta and LV cavity. Many have a history of hypertension and some have calcification of the mitral valve annulus. The limited data suggest that individuals with this pattern of ventricular remodelling are less likely to have familial disease or a mutation in a cardiac sarcomeric protein gene.488Importantly, due to provocable LVOTO, some patients with basal septal hypertrophy experience symptoms on exertion and should be assessed using physiological provocation and stress echocardiography in the same way as patients with unequivocal HCM.489,490 Advice on family screening in this group is challenging, but should be guided by the implications for family members and the presence of suspicious symptoms in relatives.
12.4 Diagnosis and management of valve disease in patients with hypertrophic cardiomyopathy
12.4.1 Aortic valve disease
In the absence of a family history of HCM or a known history of HCM before the development of significant aortic valve disease, the differential diagnosis between, on the one hand, valvular aortic stenosis with severe LVH and, on the other, HCM associated with degenerative aortic valve disease can be challenging, particularly in the elderly with hypertension. In general, the pattern and severity of LV remodelling in aortic stenosis correlates modestly with the severity of valve narrowing. Between 20% and 30% have an asymmetric pattern of wall thickening, although the severity of hypertrophy is usually relatively mild (wall thickness ≤15 mm).491,492 Left ventricular wall thickness ≥15 mm has been reported in older hypertensive patients in small cohorts, who were examined using CMR.450 Systolic anterior motion and dynamic LVOTO is reported in patients with aortic stenosis and complicates accurate measurement of the valve gradient. The treatment of aortic stenosis should be in line with current ESC Guidelines.493 In patients with aortic stenosis and in whom dynamic obstruction is not demonstrated pre-operatively, septal myectomy is controversial and not recommended for routine use.494
Up to one-third of patients with HCM have mild AR, probably caused by sub-aortic obstruction and high-velocity flow in the LV outflow tract.495,496 Moderate-to-severe AR is much less common and is usually caused by primary disease of the aortic valve leaflets or aortic root and infective endocarditis;497 when present in a patient with LVOTO, a non-SAM-related mechanism for obstruction, such as a sub-aortic membrane, should be excluded. Aortic regurgitation may also occur following septal myectomy, particularly in children and young adults.498,499 The severity of AR should be assessed in accordance with ESC guidelines by evaluating the anatomy of the valve, the size of the aortic root and of the ascending aorta, and other qualitative, semi-quantitative and quantitative parameters.500 The size of the LV cavity is an unreliable marker of the severity of AR in HCM.
12.4.2 Mitral valve disease
Mitral valve abnormalities secondary to LVOTO are discussed in section 9.1.3. The assessment of intrinsic mitral valve abnormalities can be challenging in the presence of LVOTO that, in itself, causes mitral regurgitation. The usual integrative approach to the assessment of mitral regurgitation, as recommended in the ESC/European Association for Cardio-Thoracic Surgery (EACTS) Guidelines on the management of valvular heart disease,493 has some limitations in HCM because the LV cavity is often small, even in the presence of severe mitral regurgitation, and conventional quantitative and semi-quantitative Doppler parameters are not validated in patients with LVOTO. In general, qualitative measures of valve anatomy—continuous wave and colour Doppler, combined with left atrial size and estimation of pulmonary artery pressure—are more helpful. In selected cases, TOE may be helpful in defining the mechanism and severity of mitral valve regurgitation.
12.4.3 Endocarditis prophylaxis
Infective endocarditis in HCM is virtually confined to patients with LV outflow obstruction, particularly in those with LA dilation.501 The incidence of endocarditis in patients with LVOTO was 3.8 per 1000 person-years and the probability of endocarditis 4.3% at 10 years of follow-up in a large cohort study.501 Endocardial lesions most commonly occur on the thickened anterior mitral leaflet or adjacent surface of the proximal ventricular septum.497,502 As in patients with valve disease, good oral hygiene should be encouraged, but routine antibiotic prophylaxis is not recommended in patients with LV outflow tract gradients.503 Antibiotic prophylaxis should be considered for high-risk procedures in patients with prosthetic heart valves or prosthetic material used for valve repair, previous endocarditis or congenital heart disease, in accordance with the ESC/EACTS Guidelines on the management of valvular heart disease.493,503
13. Living with cardiomyopathy: advice to patients
Most people with HCM lead normal and productive lives, but a small number experience significant symptoms and are at risk of disease-related complications. Irrespective of the severity of their disease, it is important that individuals receive support and accurate advice from family practitioners and other healthcare professionals, and that they are encouraged to understand and manage the disease themselves. Table 10summarizes some of the key issues that should be discussed with patients, relatives and carers. When appropriate (for example, when considering pregnancy), patients should be referred to other specialist services.
General lifestyle considerations for patients with hypertrophic cardiomyopathy
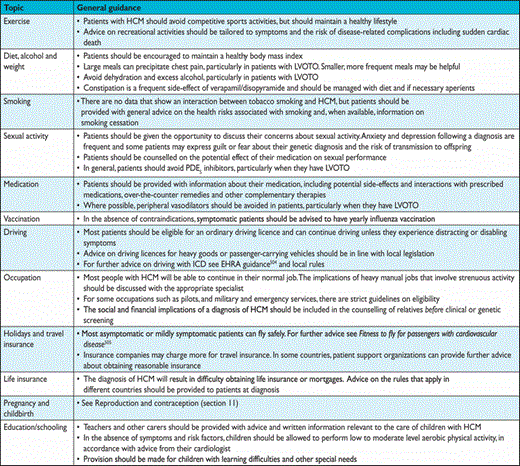 |
|
ICD = implantable cardioverter defibrillator; EHRA = European Heart Rhythm Association; HCM = hypertrophic cardiomyopathy; LVOTO = left ventricular outflow tract obstruction; PDE5 = phosphodiesterase 5.
General lifestyle considerations for patients with hypertrophic cardiomyopathy
|
|
ICD = implantable cardioverter defibrillator; EHRA = European Heart Rhythm Association; HCM = hypertrophic cardiomyopathy; LVOTO = left ventricular outflow tract obstruction; PDE5 = phosphodiesterase 5.
14. Appendix
ESC National Cardiac Societies actively involved in the review process of the 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy.
Austria:Austrian Society of Cardiology, Matthias Frick; Azerbaijan:Azerbaijan Society of Cardiology, Farid Aliyev; Belarus: Belorussian Scientific Society of Cardiologists, Svetlana Komissarova; Belgium:Belgian Society of Cardiology, Georges Mairesse; Bosnia and Herzegovina: Association of Cardiologists of Bosnia & Herzegovina, Elnur Smajić; Bulgaria: Bulgarian Society of Cardiology, Vasil Velchev; Cyprus: Cyprus Society of Cardiology, Loizos Antoniades; Czech Republic: Czech Society of Cardiology, Ales Linhart; Denmark: Danish Society of Cardiology, Henning Bundgaard; Finland: Finnish Cardiac Society, Tiina Heliö; France: French Society of Cardiology, Antoine Leenhardt; Germany: German Cardiac Society, Hugo A. Katus; Greece: Hellenic Cardiological Society, George Efthymiadis; Hungary:Hungarian Society of Cardiology, Róbert Sepp; Iceland: Icelandic Society of Cardiology, Gunnar Thor Gunnarsson; Israel: Israel Heart Society, Shemy Carasso; Kyrgyzstan:Kyrgyz Society of Cardiology, Alina Kerimkulova; Latvia: Latvian Society of Cardiology, Ginta Kamzola; Lebanon: Lebanese Society of Cardiology, Hady Skouri; Libya: Libyan Cardiac Society, Ghada Eldirsi; Lithuania:Lithuanian Society of Cardiology, Ausra Kavoliuniene; Malta: Maltese Cardiac Society, Tiziana Felice; Netherlands:Netherlands Society of Cardiology, Michelle Michels; Norway: Norwegian Society of Cardiology, Kristina Hermann Haugaa; Poland: Polish Cardiac Society, Radosław Lenarczyk; Portugal: Portuguese Society of Cardiology, Dulce Brito; Romania: Romanian Society of Cardiology, Eduard Apetrei; Russia: Russian Society of Cardiology, Leo Bokheria; Serbia: Cardiology Society of Serbia, Dragan Lovic; Slovakia: Slovak Society of Cardiology, Robert Hatala; Spain: Spanish Society of Cardiology, Pablo Garcia Pavía; Sweden: Swedish Society of Cardiology, Maria Eriksson; Switzerland: Swiss Society of Cardiology, Stéphane Noble; The Former Yugoslav Republic of Macedonia: Macedonian FYR Society of Cardiology, Elizabeta Srbinovska; Turkey: Turkish Society of Cardiology, Murat Özdemir; Ukraine: Ukrainian Association of Cardiology, Elena Nesukay; United Kingdom:British Cardiovascular Society, Neha Sekhri.