
Abstract
As a pumping organ with an intrinsic electrical system, the heart is unique. For most individuals, it remains efficient for decades. However, as a pump, it can fail. Often the failure is due to inherent or acquired problems with the electrical system. Failure of maintenance of normal sinus rhythm often results in adverse or no heart rhythm, a term referred to as “arrhythmias.” These can result in the heart rate being too fast (“tachy-”) or too slow (“brady-”) and alter blood flow resulting in patient morbidities and mortalities. Arrhythmias can occur anywhere in the heart and may not always be caused by any adverse lifestyle events such as coronary disease. Certain inherited congenital heart defects can cause abnormalities within the developing electrical system that can appear even before birth. Alternatively, the simple process of normal aging can adversely affect the heart’s ability to maintain normal rhythms. Once initiated, arrhythmias can be sustained by the normal anatomical variations of cardiac structures. There are three common arrhythmia etiologies: “automaticity” “reentry,” and “triggered.” Automaticity results from alterations of the basic cellular ion exchange mechanism which is depicted as a distinct electrical pattern, the action potential. Once an electrical impulse is initiated, it typically propagates cell to cell in a relatively uninterrupted fashion. However, if an obstruction (valves, veins) or postinfarction scar tissue occurs, the impulse can circle around the obstruction, creating a reentrant pathway. In rare instances, drugs or disease states can alter cell action potentials, triggering abnormal impulse initiation. This chapter will address all of these issues.
Similar content being viewed by others


Keywords
1 Introduction
Although perhaps somewhat of a misnomer, the term cardiac “arrhythmias” (“a” = absence of) is typically used whenever there is any alteration of normal electrical activity (rhythm) of the heart, not necessarily that there is no heart rhythm. An equally descriptive term of “dysrhythmias” (“dys” = abnormal) is also found in the literature. Both terms describe the clinical presentation of abnormal cardiac electrical impulse formation or impulse conduction through cardiac muscle. These can result in either too slow (“brady”) or too fast (“tachy”) heart rhythms. Of note, the heart is not alone in this property. Similar impulse alterations can also occur in the brain, the other primary “electrical” organ of the body. Although those brain arrhythmias are called seizures, the etiologies are comparable. As a result, pharmacotherapies can overlap.
Arrhythmias can occur at any age from the developing fetus through adulthood. Developmental alterations of the cardiac conduction tissue, genetically inherited changes of myocardial cellular ion membrane properties, and associated structural congenital heart anatomical defects can all play a role (Chap. 59). Add the normal aging process; sequelae of lifestyle habits; as well as associated metabolic disorders, systemic hypertension with resultant cardiac muscle hypertrophy, and coronary artery disease with diminished regional muscle perfusion; and an otherwise normal heart can express electrical abnormalities. However, in deference to adult-onset ischemic cardiac issues, abnormal heart rhythms occurring in young children can spontaneously disappear as the child grows. Both atrial and ventricular arrhythmias occur in the young although atrial rhythm abnormalities far exceed those of the ventricle. In the adult, atrial arrhythmias may occur solely on the basis of age, while ventricular origins are often associated with coronary blood flow alterations.
The purpose of this chapter is to inform individuals in the pharmaceutical, biomedical, as well as other health care professionals with an understanding of the “why” of arrhythmias; “what,” if any, are the predisposing factors, either structural or chemical; and the “where” in the heart they may be located. An understanding of these concepts can aid in continued future applications of selective interventions to either control or eliminate these abnormalities which, in themselves, often lead to patient morbidity and/or mortality.
2 Cardiac Electrical Development Predisposing to Arrhythmias
Intrinsically, in the development of the human heart, there exists the predisposition for arrhythmias based solely on anatomy and how that affects electrical impulse transmission, irrespective of any future ischemic cellular changes. In addition, anatomical congenital heart disease, such as septal defects or valve abnormalities, can cause alterations in the normal cardiac electrical system, predisposing to brady- or tachyarrhythmias. Any surgical repairs of these congenital defects can leave behind scars and fibrotic changes which also will predispose to eventual arrhythmias, even decades later [1, 2] (Chap. 59).
The embryogenic human heart forms as a tube and begins spontaneous peristaltic muscle contractions at approximately 23 days of gestation. The eventual specialized atrioventricular (AV) conduction system, which develops from differentiated myocytes along the coronary artery system, forms later through 6 weeks of gestation. In the majority of instances, the right coronary artery will supply blood to the sinus and AV nodes. In the region of the developing atrium near the superior vena cava-atrial junction, specialized cells form into the horseshoe-shaped sinus node at variable locations with the central area typically evolving into cells exhibiting pacemaker action potential impulse initiation. This is discussed below. Taillike projections extend into the lateral aspect of the developing atrium toward the appendage. The periphery of these tails commonly also exhibits pacemaker activity extending electrically active cell connections along the atrial wall. This anatomical arrangement can create a focus for atrial arrhythmias [3, 4]. In comparison to the developing ventricles, the atria do not contain any specialized electrical conducting tissue, per se. Although there are alignments of muscle fibers that facilitate the electrical propagation from the sinus to AV nodes (Wenckebach, Thorel) as well as right to left atrium (Bachmann), the spread of electrical impulses activity can be compared to a wave. This concept will play an important role in the initiation and propagation of such arrhythmias as atrial flutter [5].
The anatomy of the atria itself also plays a role in arrhythmias [6]. The endocardial surfaces are not smooth. As a result of embryologic connections of venous structures into the developing right and left sides, as well as anatomical landmarks permitting preferential flow patterns of placental blood, the endocardial surface is marked by irregularities, valves, openings, and ridges. Heart tissue itself is not a histologic syncytium but rather comprised of individual membrane-bound myocytes of various alignments, connected by discrete junctions. The term “cardiac anisotropy” has been applied to define these properties, all of which alter electrical flow [7]. To better understand this concept, envision standing on a beach and watching waves coming to shore. Typically, the waves are sequential, at regular intervals, and parallel. The waves die out after hitting the shore, and another follows. Now add rocks, piers, and docks and watch how the waveforms react as they bounce off these obstacles. The regular flow patterns are disrupted, and propagation is altered, resulting in waves coming back on themselves, creating a more chaotic wave distribution. If waves circle around an obstacle, they will eventually end back where they started and “reenter” the initial pathway. This constitutes the basic of many arrhythmias and helps to comprehend how, for example, atrial flutter can start and be self-sustaining. This concept will be discussed below.
As mentioned above, the initial cardiac tube has direct muscle connections between the developing atria and ventricles. With eventual AV valve formation, these direct muscle connections become disrupted. Electrical propagation is then relegated to the developing AV node and bundle of His conduction pathways, creating a single atrial-ventricular electrical conduction pathway (atrioventricular junction). This has distinct survival advantages since the specialized AV conduction system has an inherent property (decremental conduction) that slows impulse propagation from atrium to the ventricle (seen as the PR interval on a surface ECG). The ventricle is then protected from rapid and potentially fatal atrial arrhythmias (Chaps. 46 and 50).
However, in a subset of individuals, these primitive direct muscle connections remain, ostensibly bypassing the specialized conduction tissue and causing early ventricular electrical activation at the region where the fibers intersect with the ventricle. Persistence of these connections results in various “preexcitation” syndromes, such as Wolff-Parkinson-White [8, 9]. Since these muscle fibers lack the inherent capacity to slow impulses coming from the atrium, any atrial tachycardia (such as flutter) can propagate rapidly to the ventricle potentially causing sudden death. Also, instead of a single connection pathway, electrical impulses can travel along two: down one, up the other, and back down again. A reentrant circuit is created.
In the region of the apex of the anatomical triangle of Koch of the right atrium, electrophysiologic tissues, derived from cells in the sinus venous walls and atrioventricular canal regions, coalesce into a compact structure, the AV node. It varies in size, may be in the right atrial septum, across the central fibrous body, or entrapped in the tricuspid valve annulus. Rarely, it can extend leftward and even be located close to the base of the aortic valve. It is a heterogeneous structure consisting of diffuse anatomical myocellular arrangements. As an analogy, if a hairnet is pulled at one corner, the weblike strands coalesce into a more compact alignment. Previous histological studies have demonstrated comparable cellular arrangements in the para-AV node region [10]. However, as the fibers align, the muscle’s intrinsic electrical conduction properties can be altered, resulting in gap junctions with “slow” and “fast” conducting fibers exhibiting different refractory periods, allowing electrical impulses to travel between fibers. Although not necessarily associated with sudden death, these changes do permit reentrant arrhythmias associated with patient morbidities [11].
The diffuse cellular configurations at the entrance into AV node organize into a more longitudinal pattern which becomes the penetrating portion of the bundle of His. This enters into the central fibrous body and, at the crest of the interventricular septum, branches into right and left bundles. On the right, it continues along the moderator band. On the left, it rapidly spreads into a more diffuse Purkinje arrangement. In addition to structural anatomical differences between the left and right ventricles, with the left architecture more amenable to supporting higher pressures, this electrical conduction distribution pattern supports the importance of the systemic left over that of the pulmonic right ventricle. The longer course of the right bundle places that structure at increased risk for damage. This is especially true in congenital heart defects.
Any congenital anatomical defect involving the AV canal or septum can cause intrinsic errors of electrical system development as the evolving conduction tissue responds to these structural anatomical anomalies. For example, a persistent opening in the region of septum, such as seen with an AV canal or ventricular septal defect, can cause a deviation of the electrical conduction tissue away from its normal septal course, resulting in abnormal ventricular activation sequences. Intrinsic bundle branch block patterns, any degree of AV conduction delay including complete heart block, as well as an altered QRS frontal plane axis as seen on a surface ECG may, therefore, be the result of embryologic development and not necessarily always related to acquired or ischemic heart disease. This is an important concept in the evolving field of adult congenital heart disease [12, 13].
3 Electrical Impulse Initiation
The basic premise behind cardiac tissue electrical impulse initiation is the inherent ability of cells to generate an action potential (AP), either intrinsically as in the sinus or AV nodes, or in muscle cells in response to an appropriate stimulus, a concept termed “excitability.” As originally described in the 1950s in regard to nerve cells, and applicable to cardiac cells, electrolyte concentration differences associated with the continuous influx and efflux of sodium (Na++), potassium (K+), calcium (Ca++), and chloride (Cl−) ions between the interior and exterior of cells create a transmembrane electrical gradient, with an intracellular side typically −80 to −90 mV among cardiac muscle cells and −60 mV in nodal cells, with respect to the extracellular side. Potassium is the primary intra- and sodium the primary extracellular ion [14]. The typical muscle AP pattern consists of five phases of membrane ion exchange (0–4) while sinus and AV nodal cells exhibit only three (0,3,4) starting with a rapid Na++ (muscle) or slow Ca++ (nodal) influx which alters the transmembrane electrical gradient, concluding with membrane restabilization back to baseline in muscle but undergoing spontaneous depolarization back up to threshold levels in nodal cells (Fig. 47.1). This spontaneous diastolic depolarization of nodal cells maintains normal heart rates. On a surface ECG, the QRS-T components reflect these phases of the AP (Fig. 47.2) (Chap. 46).

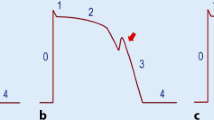
The difference between nodal (Ca++) and muscle (Na++) action potentials is illustrated. Compared with sodium channels of muscle, nodal calcium channel APs have a less negative resting membrane potential, slower upstroke, absence of phases 1 and 2. These cells inherently undergo spontaneous depolarization (automaticity) as seen by the upstroke of phase 4. Once the membrane potential reaches the threshold level, a new impulse is created

Cardiac muscle action potential with a comparable surface ECG tracing. As illustrated, AP phases are indicated (0–4) and reflect ion exchange concentrations. The ventricular QRS-T of the ECG reflects the intracellular ion flux: QR = phase 0–1, while ST-T = phases 2–3. For this reason, changes in cardiac cellular metabolism, associated with arrhythmias, will often result in ECG changes such as T wave inversion or ST elevations as a marker of intracellular abnormalities
The spontaneous membrane electrical depolarization activity, or automaticity, of nodal cells permits these cells to generate action potentials at a constant rate and act as “pacemakers,” regulating cardiac rhythm. Based on ion channel recovery duration, cells may or may not be able to propagate another AP at any given time interval, a term referred to as “refractoriness.” This property becomes important in the propagation of arrhythmias once they are initiated, and this forms a major focus in pharmacotherapeutic control of arrhythmias. The interdigitating configuration of cardiac cells permits this integrated electrical activity to spread throughout the heart, thus permitting overall muscle contraction. Detailed descriptions of these intra- and intercellular processes can be found elsewhere in numerous reports, and microelectrode voltage-clamp studies and are beyond the scope of this chapter [15]. However, a basic understanding of the normal action potential helps to put some arrhythmias into perspective since any alteration in AP duration, amplitude, or configuration can be the focus for abnormalities of cardiac rhythm (Chap. 46).
4 Altered Action Potentials Causing Arrhythmias
As mentioned above, any alterations of normal cardiac cellular action potentials either by disease, ischemia, inherited genetic conditions, cellular chemical composition, drugs, or even simply aging, can be arrhythmogenic. The sinus node becomes less efficient with age, resulting in loss of its “pacemaker” capabilities, causing slower intrinsic heart rates in the elderly but an increase in abnormal escape rhythms due to this lack of rate control. The term “sick sinus syndrome” has been applied to symptomatic arrhythmias associated with sinus bradycardia, sinus arrest, or alternating brady- and tachyarrhythmias. Since sinus nodal control of heart rate begins to decrease after the age of 55 years, the intrinsic ability of heart muscle to generate electrical activity acts against the individual with a reported nearly 25 % lifetime risk of developing atrial fibrillation based on the aging process alone [16]. Any patient with repaired congenital heart, or anyone who has been on cardiopulmonary bypass, can have resultant sinus node damage by virtue of superior vena cava cannulation or sutures. In addition, certain congenital heart defects, such as atrial septal defects, may intrinsically have abnormal sinus node function [17].
Electrolyte abnormalities can affect APs causing atrial and ventricular arrhythmias. Sodium ion overload can occur in the heart tissue resulting in Na-Ca exchange abnormalities, as seen in arrhythmias associated with tissue reperfusion following periods of ischemia. Cardiac muscle acid-base balance can also alter APs. Although a decrease in tissue pH values raises intracellular Na++ and alters contractions by an increase in Ca++ release, a severe decrease to pH 6.7 or less can be associated with Ca++ overload arrhythmias [18]. Abnormalities of the inward potassium channel (IKs), as seen with such inherited genetic conditions such as the long QT syndrome (LQT1), cause AP duration prolongation resulting in membrane instability and a predisposition to often fatal ventricular arrhythmias (polymorphic ventricular tachycardia or torsade de pointe) (Chap. 49). The APs of normal muscle cells, following any metabolic insult such as ischemic injury, can be altered, permitting spontaneous initiation of depolarization. Diseased atrial muscle fibers as well as Purkinje cells can exhibit chronic depolarizations independent of any extracellular activity. Premature atrial contractions (PACs), premature ventricular contractions (PVCs), ectopic atrial tachycardia (EAT/AET), or junctional ectopic tachycardia (JET) can be the result of these adverse membrane changes. Ischemic cells lose potassium causing an increase in extracellular K+ concentrations, which, in turn, alter transmembrane ion ratios, which adversely alter APs [19]. Calcium plays a role in delayed afterdepolarizations contributing to arrhythmias. The normally quiescent membrane ion activity during phase 4 of cardiac muscle APs can become unstable, permitting an increased inward current sufficient enough to trigger a new action potential (early afterdepolarization) and propagate what is referred to as “triggered arrhythmias.” These cellular changes can often be detected by concomitant changes in the S-T, T, and T-U wave patterns seen on the surface ECG (Chap. 48).
Pharmacologic agents, including antiarrhythmic drugs can be proarrhythmic by virtue of adverse action potential effects such as an increasing AP duration or depressing sinus or atrioventricular node activity which can allow for ectopic escape rhythms. Bradycardia contributes to torsade de pointe, while an increase in heart rate can provoke ventricular tachycardia/fibrillation. Drugs or electrolyte abnormalities (hypokalemia) that cause the AP duration to increase have been implicated in fatal arrhythmias. Excellent reviews of pharmacologic agents provoking arrhythmias can be found elsewhere [20, 21] and in Chap. 48. However, a brief listing includes antiarrhythmics (sotalol, ibutilide, and amiodarone), antibiotics (erythromycin and trimethoprin-sulfa), antihistamines (terodiline), and antiseizure agents (thioridazine and tricyclics). In addition, other drugs (psychotropic agents, antifungal agents, chemicals (organophosphates)), as well as liquid protein diets can adversely alter cardiac cell action potentials and provoke arrhythmias.
5 Reentrant Arrhythmias
As introduced above, cardiac electrical wave propagation tends to flow relatively smoothly. However, if a wave is blocked in one direction but permitted to reroute and return to the original site of excitation, a revolving pattern of wave propagation ensues: “reentry,” “circus movement.” This concept was first described in the early twentieth century [22]. This circular impulse activity can occur anywhere in the heart in which there is an impediment to electrical propagation (ischemic, anatomical, or mechanical) and is responsible for most of the important clinical tachyarrhythmias. Among these are atrioventricular reentrant tachycardia (AVRT), atrioventricular nodal tachycardia (AVNRT), atrial flutter/fibrillation, and ventricular tachycardia/fibrillation.
Initiation of a reentrant tachycardia is dependent on several factors: (1) the existence of at least two intersecting pathways or limbs (often referred to as α and β) that create a loop for the wave to travel, (2) the wave propagation must be blocked in one of the limbs of the loop and be allowed to travel down the other limb, and (3) the conduction properties of the tissue creating the loop (speed and refractoriness) must be such that the wave can continue to circle around the loop (Fig. 47.3). Although a patient may be susceptible to having a reentrant tachycardia, intrinsic tissue properties dictate when such an occurrence will happen. For example, a patient with Wolff-Parkinson-White syndrome with accessory atrioventricular pathways that form an anatomical loop with the normal AV node, may exhibit tachyarrhythmias only intermittently. Necessary conditions, often provoked by transient electrical alterations, such as premature atrial or ventricular complexes, need to occur to initiate such an arrhythmia. These reentrant circuits typically do not occur among individuals with completely normal hearts, which is why not everyone complains of abnormal tachycardias. However, even normal hearts can be made abnormal by factors that change myocellular properties.

Tissue properties that allow for a reentrant pathway to occur: 1 entering impulse, 2 obstacle (anatomy, scar, ischemic tissue, etc.), 3 impulse blocked in one limb of the loop, 4 impulse propagates with slow conduction down the other limb, 5 impulse enters up the original blocked limb in a retrograde fashion, and 6 impulse “reenters” the original limb
6 Types of Arrhythmias
The pathogenesis of the various arrhythmias has been mentioned above. As a quick review, the following should be useful in the understanding of the diverse topic of abnormal heart rhythms. It is important to remember that any region of the heart can be associated with electrical abnormalities.
-
Sinus Arrhythmia: This is more of a misnomer than anything. Heart rate variability is normal. Typically, this is due to autonomic tone and not an abnormality, per se. The normal heart rate fluctuates, often in response to physiologic changes in the body. Normal heart rate range values for patient ages are readily available elsewhere.
-
Sinus Bradycardia: This is a normal sinus rhythm but too slow for a given age. This can be due to autonomic tone, drugs, age, or sinus node damage. This may require a pacemaker if there are clinical symptoms.
-
Sinus Tachycardia: This is a normal sinus rhythm but too fast for a given age. This can be due to autonomic tone, drugs, metabolic conditions (fever, thyroid, adrenal gland), age, or sinus node damage. This may require pharmacologic agents to control. Rarely, the sinus node itself can have a reentrant tachycardia circuit (sick sinus syndrome) that can be amenable to ablation techniques.
-
Sinus Arrest/Pause: Normally, the sinus node rate fluctuates. However, due to sinus node or para-nodal tissue issues, long pauses between sinus complexes can occur. Often, these are a numerical variable of the heart rate. If symptoms develop, pacemaker therapy may be required.
-
Atrial Arrhythmia: Any abnormality of heart rhythm arising from the atria. By definition, normal sinus arrhythmia can be considered an atrial arrhythmia. However, the term is more appropriately used to categorize such heart rhythm abnormalities as premature atrial complexes, ectopic atrial tachycardias, reentrant atrial muscle tachycardias such as flutter/intra-atrial reentrant tachycardia (IART), or chaotic electrical muscle activity, and atrial fibrillation. Etiologies include normal variations as well as intrinsic heart muscle damage/disease. All three categories (automaticity, reentry, and triggered) can be causative. Single atrial complexes can be normal and seldom require any intervention, especially in infants. All others can be amenable to catheter ablation and/or pharmacologic intervention depending on clinical issues.
-
Atrial Paroxysmal Tachycardias: These are rapid atrial heart rates that come and go. These are often due to reentrant mechanisms (discussed above) associated with Wolff-Parkinson-White or other preexcitation syndromes causing atrioventricular reciprocating tachycardia (AVRT), or related to intrinsic AV nodal and para-nodal tissue configurations causing atrioventricular nodal tachycardia (AVNRT). These can be amenable to ablation or pharmacologic therapy.
-
Ventricular Arrhythmias: Comparable to abnormalities of heart rhythm arising from the atria, the ventricles can also be the source for arrhythmias. However, these typically are more clinically significant since they can be associated with higher patient mortalities. Again, most are related to rapid reentrant mechanisms (ventricular tachycardia) which, however, can degenerate into uncontrolled rhythms (ventricular fibrillation, torsade de pointes) as a prodrome to sudden death. Etiologies include inherited cardiac muscle disease (hypertrophic cardiomyopathy), inherited cardiac electrical disease (long QT syndrome), and ischemic heart muscle due to coronary disease, surgical scars, and drugs. Single premature complexes (PVCs) can be related to all of the above etiologies or can be normal, especially in adolescents (benign ectopy of adolescents). Clinical correlation is mandatory in all situations.
-
Heart Block: As discussed above, congenital developmental abnormalities of cardiac structures, ischemic muscle damage, drugs, disease, or surgical scars can adversely affect the developing electrical conduction system. Delays in impulse propagation through the atrioventricular conduction system can result in slow conduction (first-degree AV block) detected by a prolongation of the PR interval on the resting EKG, intermittent conduction (second-degree AV block), or no conduction (third-degree (complete) AV block). Impulse delay further in the ventricles can result in either a right or left bundle branch block. In the former conditions affecting the AV node, symptoms of bradycardia, including Stokes-Adams attacks, may require pacemaker therapy. In the latter ventricular conditions, ventricles are depolarized abnormally resulting in ventricle contractility issues.
7 Concluding Remarks
As an electrical organ, the heart initiates and sustains impulse formation and propagation. Typically, it functions appropriately. However, normal anatomical configurations, congenital defects of development as well as the basic cellular matrix predispose all hearts, even among individuals with no prior history of cardiac disease, to a susceptibility to arrhythmias. Add ischemic cellular changes associated with lifestyle, other disease states, and aging, and this susceptibility increases. This chapter has discussed etiologies, locations, and some clinical correlations of the various types of cardiac rhythm abnormalities to which anyone in the health care field will be exposed. An understanding of normal anatomy or congenitally defective or inherited cardiac conditions as well as altered disease states, as they predispose and contribute to cardiac arrhythmogenesis, enables future study into how to effectively treat and potentially prevent associated morbidities and mortalities associated with adverse heart rhythms.
References
Webb CL, Jenkins KJ, Karpawich PP, Bolger A, Donner RM, Allen HD, Barst RJ, Committee on Congenital Heart Defects, Section of Cardiovascular Disease in the Young, American Heart Association. Collaborative care for adults with congenital heart disease. Circulation. 2002;105(19):2318–23.
Nakagawa H, Shah N, Matsudaira K, Overholt E, Chandrasekaran K, Beckman KJ, Spector P, Calame JD, Rao A, Hasdemir C, Otomo K, Wang Z, Lazzara R, Jackman WM. Characteristics of reentrant circuit in macroreentrant right atria tachycardia after surgical repair of congenital heart disease. Circulation. 2001;103:699–709.
Wu D. Demonstration of sustained sinus and atrial reentry as a mechanism of paroxysmal supraventricular tachycardia. Circulation. 1975;51:234–43.
Nakagawa H, Lazzara R, Khastgir T. Role of the tricuspid valve and the Eustachian valve/ridge on atrial flutter. Circulation. 1996;94:407–24.
Ho SY, Anderson RH, Sanchez-Quintana D. Atrial structures and fibres: morphologic bases of atrial conductance. Cardiovasc Res. 2002;54:325–36.
Valderrabano M. Influence of anisotropic conduction properties in the propagation of the cardiac action potential. Prog Biophys Mol Biol. 2007;94:144–68.
Wolff L, Parkinson J, White P. Bundle branch block with short P-R interval in healthy young people prone to paroxysmal tachycardia. Am Heart J. 1930;5:685–704.
Anderson RH, Becker AE. Accessory atrioventricular connections. J Thorac Cardiovasc Surg. 1979;78:310–1.
Anderson R, Becker A, Brechenmacher C, Davies MJ, Rossi L. The human atrioventricular junctional area, a morphological study of the AV node and bundle. Eur J Cardiol. 1975;3:11–25.
Nikolski VP, Jones SA, Lancaster MK, Boyett MR, Efimov IR. Cx43 and the dual-pathway electrophysiology of the atrioventricular node and atrioventricular nodal reentry. Circ Res. 2003;92:469–75.
Walsh E, Cecchin F. Arrhythmias in adult patients with congenital heart disease. Circulation. 2007;115:534–45.
Khairy P, Van Hare GF, Balaji S, Berul CI, Cecchin F, Cohen MI, Daniels CJ, Deal BJ, Dearani JA, Groot ND, Dubin AM, Harris L, Janousek J, Kanter RK, Karpawich PP, Perry JC, Seslar SP, Shah MJ, Silka MJ, Triedman JK, Walsh EP, Warnes CA. PACES/HRS expert consensus statement on the recognition and management of arrhythmias in adult congenital heart disease. Heart Rhythm. 2014. doi:10.1016/j.hrthm.2014.05.009. pii:S1547-5271(14)00513-X, [Epub ahead of print].
Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–44.
Zipes D, Jalife J, editors. Cardiac electrophysiology: from cell to bedside. 4th ed. Philadelphia: Saunders; 2004.
Heeringa J, van der Kuip D, Hofman A, Kors J, van Herpen G, Stricker B, Stijnen T, Lip G, Witteman J. Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur Heart J. 2006;27:949–53.
Karpawich PP, Antillon JR, Cappola PR, Agarwal KC. Pre- and Postoperative electrophysiologic assessment of children with secundum a trial septal defect. Am J Cardiol. 1985;55:519–21.
Schwartz A, Noble D. Cardiac ion pumps and ion exchangers. In: Spooner P, Rosen M, editors. Foundations of cardiac arrhythmias: basic concepts and clinical approaches. New York: Markel Decker; 2001.
Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–169.
Rosen M, Janse M, Wit A, editors. Cardiac electrophysiology: a textbook. New York: Futura Publishing; 1990.
Slater W, Lampert S, Podrid P, Lown B. Clinical predictors of arrhythmia worsening by antiarrhythmic drugs. Am J Cardiol. 1988;61:349–53.
Mines GR. On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can. 1914;IV:43–52.
Frame L, Bernstein R. Reentry in clinical arrhythmias. In: Rosen M, Janse M, Wit A, editors. Cardiac electrophysiology: a textbook. New York: Futura Publishing; 1990.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Karpawich, P.P. (2015). Pathophysiology of Cardiac Arrhythmias: Arrhythmogenesis and Types of Arrhythmias. In: Jagadeesh, G., Balakumar, P., Maung-U, K. (eds) Pathophysiology and Pharmacotherapy of Cardiovascular Disease. Adis, Cham. https://doi.org/10.1007/978-3-319-15961-4_47
Download citation
DOI: https://doi.org/10.1007/978-3-319-15961-4_47
Publisher Name: Adis, Cham
Print ISBN: 978-3-319-15960-7
Online ISBN: 978-3-319-15961-4
eBook Packages: MedicineMedicine (R0)