


Introduction to Sanger sequencing
Sanger sequencing, developed by Frederick Sanger and colleagues in 1977, is a pioneering method for DNA sequencing. This technique relies on electrophoresis and involves the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. Initially, it was the predominant sequencing method for four decades, first commercialized by Applied Biosystems in 1986.
- The first direct DNA sequencing method
- Sequencing by synthesis
- Di-deoxy termination method
- DNA polymerase + Pool of unmodified dNTPs + Pool of di-deoxy NTPs
Methodology
It uses chain-termination method which requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotide triphosphates (dNTPs), and modified di-deoxynucleotide triphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3′-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines.
What is the difference between a deoxyribonucleotide and a dideoxyribonucleotide? Why dideoxyribonucleotide is used in Sanger’s method of DNA sequencing? What will happen if very high or very low amount of ddNTPs are used in Sanger’s method of DNA sequencing? Discuss.
The key difference between a deoxyribonucleotide and a dideoxyribonucleotide lies in their chemical structure. A deoxyribonucleotide contains a deoxyribose sugar molecule, a nitrogenous base (adenine, guanine, cytosine, or thymine), and a phosphate group. On the other hand, a dideoxyribonucleotide lacks the 3′ hydroxyl group on the deoxyribose sugar, making it incapable of forming further phosphodiester bonds.
Dideoxyribonucleotides are used in Sanger’s method of DNA sequencing as chain terminators. When a dideoxyribonucleotide is incorporated into a growing DNA strand during the sequencing reaction, it prevents further elongation of that strand, resulting in DNA fragments of varying lengths.
| Property | Deoxyribonucleotide | Dideoxyribonucleotide |
|---|---|---|
| 3′ Hydroxyl Group | Present | Absent |
| Forms Phosphodiester Bonds | Yes | No |
| Function in Sanger Sequencing | Allows strand elongation | Terminates strand elongation |
 |  |
If very high amounts of ddNTPs (dideoxyribonucleotides) are used in Sanger’s method:
- More DNA strands will terminate prematurely
- Resulting in shorter fragment lengths
- Potentially leading to incomplete or inaccurate sequence information
If very low amounts of ddNTPs are used:
- Fewer DNA strands will terminate
- Resulting in longer fragment lengths
- Potentially making it difficult to resolve and distinguish individual fragments
- Leading to incomplete or inaccurate sequence information
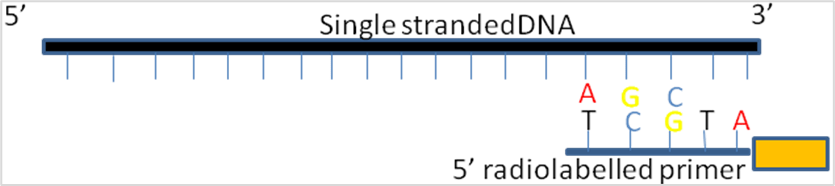

The Sanger (chain-termination) method for DNA sequencing.

- The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase.
- To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones.
- The deoxynucleotide concentration should be approximately 100-fold higher than that of the corresponding dideoxynucleotide (e.g. 0.5mM dTTP: 0.005mM ddTTP) to allow enough fragments to be produced while still transcribing the complete sequence
- Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis.
- The DNA bands can be visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image.
DNA Sequence Analysis by Autoradiograph
After the completion of each reaction, Polyacrylamide gel electrophoresis (PAGE) is performed. Each reaction mix is loaded in a separate lane. The reaction condition should be carefully controlled to separate the strands that differ just by a single nucleotide. PAGE is done in denaturating condition in presence of urea or less frequent formamide. Urea and formamide lowers the melting point of DNA molecule, denatures DNA by disrupting the H bond and the newly synthesized strand separates from the template strand.
Electrophoresis is carried out at high voltage to prevent the renaturation of DNA due to high heat generation in gel.
After complete run, the gel is transferred on nitrocellulose filter and autoradiography is performed so the only bands having the 5’ radiolabelled molecule will be visible as bands. In PAGE the shortest fragment moves faster so the bottom most molecule is the first dideoxynucleotide which stopped the chain elongation by its incorporation and thus that should be the first sequenced nucleotide. So by this bottom up approach in all the lanes we can get a combined DNA sequence of the query (Fig .4).


Applications of Sanger Sequencing
- Sanger sequencing is still widely used for smaller-scale projects, where its accuracy and longer read lengths are beneficial.
- It is employed for the validation of results obtained from deep sequencing technologies.
- Active use in public health initiatives, such as sequencing the spike protein from SARS-CoV-2.
- Utilized by the CDC’s CaliciNet surveillance network for monitoring norovirus outbreaks.
Key differences between Sanger sequencing and NGS
| Feature | Sanger Sequencing | Next-Generation Sequencing (NGS) |
| Principle | Chain-termination method | Parallel sequencing of millions of DNA fragments |
| Development | Introduced in 1977 by Frederick Sanger | Emerged in the mid-2000s |
| Read length | > 500 nucleotides (long reads) | Short reads (100s of base pairs), with some platforms achieving longer reads |
| Accuracy | Very high (around 99.99%) | High, but may vary between platforms |
| Speed | Slower | Faster, capable of high-throughput sequencing |
| Cost per Bae | Higher cost per base | Lower cost per base |
| Applications | Often used for smaller-scale projects, validation of results | Commonly used for large-scale genome analysis, whole-genome sequencing, transcriptomics, etc. |
| Instrumentation | Requires capillary electrophoresis machines | Requires specialized NGS platforms |
| Labour intenstity | Labor-intensive, manual processes | Less labor-intensive due to automation |
| Error rate | Low error rate (high accuracy) | Low, but may vary by platform and read length |
| Use in public health | Active role in initiatives like sequencing the spike protein of SARS-CoV-2, surveillance of norovirus outbreaks through CDC’s CaliciNet | Commonly used in various public health initiatives due to high-throughput capabilities and rapid results |
Human Genome Sequencing Project
- Too expensive and time consuming!
- Completed in 2003, took ~13 years
- Cost: USD 3 billion

Drawbacks of Sanger sequencing method
- Read Length Limitation:
- Sanger sequencing is limited in its ability to generate long reads compared to some modern sequencing technologies. Read lengths are typically limited to around 500-800 nucleotides.
- Low Throughput
- Sanger sequencing is a relatively low-throughput method. It involves separate reactions for each DNA fragment, making it less suitable for large-scale or high-throughput sequencing projects.
- Labor-Intensive
- The process of Sanger sequencing involves manual steps, such as gel preparation, loading, and analysis. This makes it more labor-intensive compared to automated, high-throughput methods like NGS.
- Cost-Per-Base
- Sanger sequencing can be more expensive on a per-base basis, especially for longer sequences. The cost per base can become a limiting factor for large-scale sequencing projects.
- Limited Multiplexing
- Multiplexing, the simultaneous sequencing of multiple samples in a single run, is limited in Sanger sequencing. NGS technologies offer much higher multiplexing capabilities.
- Inability to Discriminate Homopolymeric Regions
- Sanger sequencing may have challenges in accurately determining the length of homopolymeric regions (repeating nucleotides) due to the difficulty in resolving them on the sequencing gel.
- Not Suitable for Metagenomics
- Sanger sequencing is less suitable for metagenomic studies, where the goal is to sequence genetic material from multiple species within a complex sample. NGS is more adept at handling such diversity.
- Slower Turnaround Time
- Sanger sequencing, with its manual steps and longer individual reaction times, usually has a slower turnaround time compared to NGS methods.
- Limited Dynamic Range
- Sanger sequencing may face challenges in accurately quantifying variations in DNA abundance over a wide dynamic range, especially in complex samples.
While Sanger sequencing continues to be valuable for certain applications, especially those requiring high accuracy for short sequences, its limitations have led to the widespread adoption of NGS technologies for large-scale genomics projects.
Emergence of Next Generation Sequencing (NGS) Technologies
Needed:
- Direct sequencing method
- high-throughput, accurate and reproducible
- cost-effective
Target: Human genome sequencing for $1000

Benefits of NGS Technologies
- Ability to sequence thousands of genes or genomic regions simultaneously
- Ability to directly sequence unknown genomic fragments or genomes
- Capability to sequence a large number of samples in a short time
- More power to detect low frequency variants
- Cost-effective for processing a large number of samples

