
Abstract
EuGene is an integrative gene finder applicable to both prokaryotic and eukaryotic genomes. EuGene annotated its first genome in 1999. Starting from genomic DNA sequences representing a complete genome, EuGene is able to predict the major transcript units in the genome from a variety of sources of information: statistical information, similarities with known transcripts and proteins, but also any GFF3 structured information supporting the presence or absence of specific types of elements. EuGene has been used to find genes in the plants Arabidopsis thaliana, Medicago truncatula, and Theobroma cacao; tomato, sunflower, and Rosa genomes; and in the nematode Meloidogyne incognita genome, among many others. The large fraction of plant in this list probably influenced EuGene development, especially in its capacities to withstand a genome with a large number of repeated regions and transposable elements.
Depending on the sources of information used for prediction, EuGene can be considered as purely ab initio, purely similarity based, or hybrid. With the general availability of NGS-transcribed sequence data in genome projects, EuGene adopts a default hybrid behavior that strongly relies on similarity information. Initially targeted at eukaryotic genomes, EuGene has also been extended to offer integrative gene prediction for bacteria, allowing for richer and robust predictions than either purely statistical or homology-based prokaryotic gene finders.
This text has been written as a practical guide that will give you the capacity to train and execute EuGene on your favorite eukaryotic genome. As the prokaryotic case is simpler and has already been described, only the main differences with the eukaryotic version were reported.
Similar content being viewed by others

Key words
1 Introduction
1.1 What Is EuGene
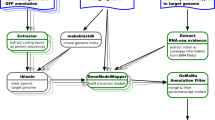
EuGene is a machine learning-based integrative gene finder. It aims at annotating a complete genome sequence with a precise delineation of likely transcribed or translated regions on each strand of the input genome. It is therefore not limited to the prediction of protein-coding exons and introns but can also predict UTRs and ncRNA genes. To maximize the quality of the prediction, EuGene has been designed to easily incorporate a variety of information. It immediately departed from the pure ab initio HMM-based approach that exclusively relies on statistical sequence information to also incorporate multiple features that are directly related to the annotation. Each type of information can be integrated in a modular software architecture based on the use of plug-ins. In its current standard usage, EuGene incorporates:
-
Probabilistic models of intergenic, intronic, transcribed, and translated regions and their boundaries such as splice sites. These models are typically automatically trained for every new genome but can also be reused from models that have been trained on a family of genomes (group of related species).
-
Information extracted from alignment/mapping of sequenced transcripts (RNA-seq, short/long or assembled transcripts) that are considered as evidence of transcription (be it for ncRNA or protein genes) and (possibly non-canonical) splicing.
-
Information on similarities with known protein sequences which is considered as an indication of protein-coding regions.
-
Information on repeats identified by dedicated repeat detection tools. These regions are automatically delineated and masked for prediction but can be locally unmasked if similarities to a transcriptome or proteome exist.
-
Output of existing specialized genome analysis tools that can be informative on various region types (e.g., the output of specialized gene finders for ncRNA genes are directly integrated by EuGene).
With the general availability of NGS-transcribed sequence data in genome projects, EuGene now adopts a default hybrid behavior that exploits similarities with expressed sequences and proteins to automatically build training sets and also to perform its prediction on the whole genome. Because of its wide availability and high quality and quantity, RNA-seq has replaced several previously used types of information (e.g., conserved regions between different genomes [1]). Thanks to a very general plug-in named “AnnotaStruct”, EuGene can integrate various GFF3 information on gene structures. This makes it capable of integrating new or unusual data types very easily. This plug-in is used, for example, to integrate the output of the ncRNA gene predictors tRNAscan-SE [2], RNAmmer [3], and cmsearch on Rfam [4].
None of these sources of information is considered as absolutely certain inside EuGene. Each source of information is instead weighted before it is integrated in the prediction process. This weighting is parameterized by few (typically one or two) parameters. These parameters are pre-set to values that have previously been optimized on a group of species and can directly be used for a new species of the same group.
1.2 How Does EuGene Work
EuGene has been designed using a pure optimization approach: given a genomic DNA sequence to annotate, a specific weighted directed acyclic graph that can represent every possible annotation of the sequence is built, edited, and weighted on the fly in such a way that a constrained maximum weight path in this graph defines a consistent prediction of optimal quality [5, 6]. The edge editing and weighting process is performed by the plug-ins, each representing a source of information. Every plug-in outputs raw votes (real numbers) on specific types of edges that represent region types at every base of the genomic sequence or specific sites like splicing or transcription and translation start/stop between bases. These votes are then transformed by a parameterized scaling function to contribute to the weight of every edge. As an example, the “Stop” plug-in exploits the existence of a STOP codon in the genomic sequence to edit the graph and disable all predictions that would generate an in-frame STOP codon and instead force them to become a 3′UTR. Similarly, the “BlastX” plug-in will transform the existence of a similarity with a protein into votes in favor of a coding region in the corresponding region/frame.
The parameters of the statistical voters require a sequence training set of each predicted type (coding, transcribed but non-coding, etc.) to produce their raw votes. This learning set is automatically built by the EuGene pipeline from a stringent selection of similarities between the genomic sequence, a reference dataset of proteins and assembled transcripts of the same species. The parameters of the statistical models (Markovian models) are estimated using a maximum likelihood criterion.
To balance the strength of all raw votes, EuGene uses scaling parameters that have previously been optimized on various genomes. These parameters do not need to be re-optimized in the general case, even if this is possible in practice. They have initially been set by maximizing the empirical quality of the prediction (a linear combination of the fractions of proper and improper predictions, at the base, exon/intron, and gene level) on sets of annotated sequences. This was achieved using a combination of a genetic algorithm and a block coordinate descent algorithm.
Once properly trained and weighted, the voters are able to edit and weight the graph. An optimal (longest) path in the graph, representing the prediction that accumulates more votes than any other, is obtained using a dedicated dynamic programming algorithm (a sophisticated variant of the Bellman algorithm [7], similar to the Semi-HMM Viterbi algorithm), that takes into account the lengths of the regions in the prediction.
For mathematically oriented readers, EuGene can also be presented as a Semi Linear Conditional Random Field (see [8] for such a description), although it was designed and implemented before this mathematical model was known and published [9].
The topology of the graph (the gene model) used by EuGene is obviously different in the eukaryotic and prokaryotic mode. Compared to the eukaryotic mode that knows about splicing, the prokaryotic mode instead allows for overlapping coding genes and operons (polycistronic transcripts). EuGene can be run on both strands simultaneously or independently on each strand. In the latter case (default for the prokaryotic mode), there is no constraint in terms of overlapping predicted genes (coding or not) which allows for the prediction of, e.g., antisense ncRNA genes, when transcriptional evidence exists.
1.3 EuGene in Practice
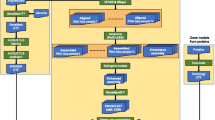
The EuGene software is the combination of a Perl pipeline that is in charge of organizing and collecting all the evidences, and C++ programs that actually perform the training and prediction. After intensive usage on a variety of eukaryotic genomes of various sizes and complexities (e.g., the Helianthus annuus L. genome has 3.5 Gb and a very high complexity [10]), the complete EuGene pipeline has evolved to be reliable, easily configurable, and suitable for various computational architectures:
-
It starts from a single directory containing the genomic sequence to annotate and produces a GFF3-formatted annotation as well as additional files in FASTA format for the predicted transcriptome, proteome, etc. in another new directory.
-
All the execution is controlled from a single configuration file, in text format, that specifies the transcriptomes and proteomes, which should be used for training and prediction, as well as few specific global options. In its default mode, the eukaryotic configuration file expects a transcriptome defined by assembled transcripts. The intronless prokaryotic version directly uses stranded NGS reads.
-
The program can exploit the capacities of multi-core machines or clusters. Various schedulers (e.g., PBS, SGE, SLURM) are supported. The usage of a cluster is advised for the annotation of large eukaryotic genomes (more than few hundreds Mb) because of the computational cost of protein similarity identification (which is internally optimized for large genomes using a sliding window).
-
The pipeline also allows for warm restarts following unexpected interruptions or the addition or removal of informative transcriptomes and proteomes.
2 Setting Up an EuGene Pipeline Instance
In this section, we describe the installation and use of the eukaryotic version of the pipeline under Linux. In the following command lines, the symbol “%” represents the shell prompt and assumes that a “sh” shell (such as bash) is used. If you use a “csh” shell, replace the export commands with setenv. Use the text editor of your choice instead of vi.
2.1 Installation and Compilation
Download the 1.5 release of the Eukaryotic EuGene pipeline available here: http://eugene.toulouse.inra.fr/Downloads/egnep-Linux-x86_64.1.5.tar.gz. Uncompress the tarball in the directory where you want to install EuGene, and run the installation script for all the software dependencies described in Table 1:
% gzip -cd egnep-Linux-x86_64.1.5.tar.gz | tar xvf – % cd egnep-1.5 % sh sh.make_install.euk.bash
Download and install RNAmmer from http://www.cbs.dtu.dk/services/RNAmmer. It is free for academics once the license has been read and accepted. Note that EuGene can work without it: in this case, only the ribosomal RNAs detected by Rfam will be integrated in the prediction (see Subheading 3.6).
Check that the necessary Perl modules for the pipeline are installed by running the following command until it displays “Required perl modules found”:
% $PWD/bin/int/check_requirements.pl
Define the EGNEP and EUGENEDIR environment variables that respectively hold the Perl EuGene pipeline directory and the EuGene C ++ software directory. By default, the latter is installed in the parent directory of the pipeline directory. You must use absolute paths. These two variables must be set at installation and each time the pipeline is used.
% export EGNEP=$PWD % export EUGENEDIR=$EGNEP/../eugene-4.2a
2.2 Resource Requirements and Execution Time
EuGene can be executed in a multi-core/cluster environment using a Perl software (paraloop) compatible with multicore systems, SGE, SLURM, or PBS clusters. Choose the appropriate configuration file according to your system and adapt it. In the following example, we work with a SGE cluster, and the jobs are submitted to the queue named main.q:
% cd $EGNEP/bin/ext/paraloop/etc % cp templates/paraloop.root.cfg_SGE paraloop.root.cfg % vi paraloop.root.cfg PARALOOP_queue=main.q PARALOOP_qsub_params=
In most environments, the PARALOOP_*_params key has to be uncommented and initialized according to the resources of cluster nodes (e.g., memory).
The running time depends on multiple factors. It depends on the genome size and complexity, the size and the number of datasets provided as sources of evidence, and obviously on the resources of the computing environment (number of nodes available for the user, processor performances). On a cluster of 500 cores (without concurrency on the resources), one can estimate the running time (elapsed) to a day for a fungus genome, less than 1 week for a plant genome of 500 Mb, and less than 2 weeks for a complex plant genome like sunflower (>3 Gb). Note that the running time has been optimized for large sequences. Consequently, it is preferable to build the pseudomolecules before running the annotation (vs. running the annotation on contigs prior scaffolding).
2.3 EuGene Execution
To use the EuGene pipeline, you need to create three directories corresponding to the script parameters indir, outdir, and workingdir and run the main script. indir contains the genomic sequences that you want to annotate. outdir is the directory where the results of the annotation are saved. workingdir is the directory where the pipeline writes all temporary files.
Once the directories are created, copy the multi-FASTA files of the genomic sequences to annotate in indir. There is no constraint on the extension of file names. Files can be compressed with gzip, bzip2, xz, or lzma. Once the configuration file has been completed (see Subheading 3.1), the use of the eukaryotic variant of EuGene consists of a single command line with four parameters:
% $EGNEP/bin/int/egn-euk.pl \ --indir /path/of/myindir \ --outdir /path/of/myoutdir \ --workingdir /path/of/myworkingdir \ --cfg /path/of/myeugeneconffile.cfg
3 Performing a Genome Annotation with EuGene
Before running the pipeline, we advise you to check that the headers of all your multi-FASTA files (genome, transcriptomes, proteomes) do not contain any special characters (such as “|”). Keep in mind that you always need to use absolute paths in the configuration file and in the command lines.
3.1 A Single Configuration File
A single configuration file centralizes all the information EuGene needs to annotate a genome: the user-supplied data (transcriptomes, proteomes, complementary results), the paths and parameters of the external programs, and the EuGene-specific parameters. A configuration file describes a “recipe,” i.e., all the ingredients (datasets used as sources of evidence) and all the measurements (cutoffs, filters, weights) that are useful and needed for annotating a genomic sequence. Configuration files that have been used recently to annotate complete genomes (e.g., that of the Medicago genome) are available on the page http://eugene.toulouse.inra.fr/Configuration.
Each line is of the form key = value. Lines beginning with “#” are comments. The first part of the file must be filled in: this is where the user describes the ingredients of his recipe. All the parameters of the second part have default values, which should be modified with caution. The symbols “%i” and “%e” mean $EGNEP and $EUGENEDIR, respectively. This abbreviated notation makes it possible to make the names of certain paths shorter when the referred files are stored in these directory trees.
Create a copy of the generic configuration file:
% cp $EGNEP/cfg/egnep.cfg $EGNEP/cfg/egnep.myspecies.recipe1.cfg % vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg
In your configuration file, fill in:
-
1.
The name of your organism, without spaces:
organism=myspecies
-
2.
The prefix of the output files, without spaces. All result files will start with this string:
output_prefix=myspecies.20180901
-
3.
The path of the RNAmmer software (result of the command "% which rnammer").
prg_rnammer=/path/to/rnammer/exe
After you register at https://www.girinst.org/repbase, download RepBaseXX.XX_REPET.embl.tar.gz tarball into the $EGNEP/db directory. Untar it, and then specify the path of the repbaseXX.XX_aaSeq_cleaned_TE.fa file in your configuration file (XX.XX string must be replaced by the downloaded version of RepBase):
% cd $EGNEP/db/ % gzip -cd RepBaseXX.XX_REPET.embl.tar.gz | tar xvf - % vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg
repeat_sequence_db=%i/db/RepBaseXX.XX_REPET.embl/repbaseXX.XX_aaSeq_cleaned_TE.fa
3.2 Evidence Datasets
This section describes how to define the transcriptomes and proteomes that you want to integrate into your recipe to guide the prediction. The result of the annotation process depends directly on the quality and completeness of transcriptomes and proteomes, particularly those used to train statistical models. Indeed, for each new genome, probabilistic models are automatically trained from a stringent selection of similarities with a so-called “training” transcriptome and proteome.
3.2.1 Transcriptomes
The evolution of sequencing technologies has transformed the number and type of transcribed sequences produced: a decade ago, ESTs (expressed sequence tags) were generated; for several years, RNA-seq has been producing shorter reads, but in much greater quantities; thanks to new Iso-Seq (isoform sequencing) applications, it is now possible to directly sequence full-length transcripts, which in a few years should be the standard in the production of expressed sequences. To be able to integrate these different types of sequences with the same protocol, the RNA-seq data must be assembled into transcripts before being used inside the EuGene pipeline. Note that some transcriptome assemblers (e.g., Trinity) generate many partially matured (intron-retaining) transcripts. For a given locus of the Trinity output, we recommend to keep only the contig with the longest ORF which is likely the corrected matured transcript. The use of raw Trinity files can generate an important number of fragmented gene models.
The alignments of transcripts on the genome strongly guide the gene models predicted by EuGene, so it is essential to have at least one good quality transcriptome of the species to be annotated or of a very close genotype/strain/race. The training transcriptome (only one must be chosen) must be of the species to be annotated or of a very close genotype.
In practice:
Copy the transcriptomes you want to integrate to a directory (e.g., $EGNEP/db/), and edit the configuration file. For each transcriptome, add a block of six lines. Below is an example that describes a transcriptome X (X must be a number):
% vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg est_X_file=/path/of/my/transcriptomeX.fasta est_X_pcs=30 est_X_pci=97 est_X_remove_unspliced=0 est_X_training=1 est_X_preserve=1
Parameter details:
-
est_X_file. Full path of the multi-FASTA file.
-
est_X_pcs and est_X_pci (value between 0 and 100). The mapping software performs a spliced alignment of the transcripts against the genomic sequence. Transcripts that align with an alignment that spans more than est_X_pcs percents of the transcript length with a sequence identity that is greater than est_X_pci are retained. For each transcript, only the best GMAP alignment is retained.
-
est_X_remove_unspliced (possible values: 0, 1, 2). Beyond sequence similarity, a spliced alignment necessarily implies the presence of two splice sites at the endpoints of each intron. Intuitively, the presence of these signals in the genomic sequence gives a higher confidence in the alignment when it exists. The parameter est_X_remove_unspliced allows one to give a different weight to spliced alignments: if the value is 0, we give the same weight to all alignments; if its value is 1, the unspliced alignments are ignored; if its value is 2, more weight is given to the spliced alignments.
-
est_X_training (possible values: 0, 1). The value 1 makes it possible to identify the training transcriptome (a single training dataset is expected).
-
est_X_preserve (possible values: 0, 1). The value 1 means that the genomic region where a transcript aligns is “protected” against repeat masking (see Subheading 3.5).
Then, provide a list of the transcriptome numbers to be included in the recipe. Below is an example to integrate the transcriptomes X, U, and V (Configured as described above):
% vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg est_list=X U V
-
est_list. Non-empty list of transcriptome numbers to use in the recipe (the order of numbers does not matter). The list must contain a training transcriptome.
To build the training set for the generation of statistical models, EuGene stringently filters the alignments of the training transcriptome. The threshold values applied are defined by the following parameters (it is not recommended to modify them):
training_est_pcs=99 training_est_pci=99 training_est_remove_unspliced=1
The more one integrates transcriptomes from different organs or tissues and/or the greater the depth of sequencing, the more different splice variants or partially matured messenger RNAs (with intron retention) will be found in the assemblies. In some cases, local alignments are inconsistent with each other. So, only alignments containing predominantly represented introns are preserved by default. To disable this option, change the gmap_intron_filter key to 0 in the configuration file.
EuGene automatically removes small exons at the ends of transcript alignments. If you want to change this behavior, two values must be edited in the configuration file: “trim-end-exons” value in the prg_gmap_param parameter and “minlen” value in the prg_gmap_filter_patch parameter. Here is an example to indicate a minimum length of 30 nt:
% vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg prg_gmap_param=-n0 -B 5 -t 16 -L 100000 -K 25000 --trim-end-exons= 30 prg_gmap_filter_patch=%i/bin/int/misc/filter_transcript_smallendexons.pl –minlen 30
3.2.2 Proteomes
EuGene can use similarities with known proteins from species which are more or less curated and/or similar to the one we want to annotate. To this end, choose one or more proteomes; we advise to use at least the public and curated UniProtKB/Swiss-Prot database, and possibly the taxonomic division of trEMBL corresponding to your species. The training proteome (only one must be chosen) must be as complete and curated as possible. In the absence of the proteome of a closely related species, choose UniProtKB/Swiss-Prot.
In practice:
Copy the multi-FASTA proteomes to a directory (e.g., $EGNEP/db/), and then index them individually with the following command:
% $EGNEP/bin/ext/ncbi-blast/bin/makeblastdb -in /path/of/my/proteomeY.fasta \ -dbtype prot -parse_seqids
The next step is to describe the integration of the proteomes into the configuration file. For each proteome, add a block of six lines. Below is an example that shows how to describe a proteome Y (Y must be a number):
% vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg blastx_db_Y_file=/path/of/my/proteomeY.fasta blastx_db_Y_weight=0.3 blastx_db_Y_pcs=50 blastx_db_Y_remove_repet=0 blastx_db_Y_preserve=1 blastx_db_Y_training=1
Parameter details:
-
blastx_db_Y_file. Full path of the multi-FASTA file.
-
blastx_db_Y_weight (a decimal number). Confidence in the proteome. We advise to choose a value between 0.1 (e.g., for trEMBL) and 0.5 (for a proteome of a well-annotated close species).
-
blastx_db_Y_pcs (value between 0 and 100). Minimum percentage of alignment of the protein length.
-
blastx_db_Y_remove_repet (possible values: 0, 1). If the value is 1, proteins that have a similarity to a protein associated with repeats are deleted (see Subheading 3.5). We suggest a value of 1 to limit the prediction of proteins related to transposable elements.
-
blastx_db_Y_preserve (possible values: 0, 1). The value 1 means that the genomic regions where a protein similarity is present are “protected” against repeat masking (see Subheading 3.5).
-
blastx_db_Y_training (possible values: 0, 1). The value 1 makes it possible to identify the training proteome (a single protein training dataset is expected).
Then list the numbers of the proteomes to be included in the recipe. Below is an example to integrate the proteomes A and C (configured as described above):
% vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg blastx_db_list=A C
-
blastx_db_list. Non-empty list of proteome numbers to use in the recipe. The list must contain a training proteome.
From here, your configuration file is complete. The next step is to adapt EuGene to your data and genome characteristics, to change default settings or to activate certain options.
3.3 Splice Sites
3.3.1 Statistical Models for the Detection of Splice Sites
Eugene uses already trained statistical models (as WAM matrix [11]) to capture the signals contained in the regions around the splice sites. The default matrix downloaded at installation is suitable for dicot plant genomes. There are other pre-trained matrices for other species groups, including nematodes, oomycetes, and fungi. You can consult the available matrices and the species used to build them on the web page http://eugene.toulouse.inra.fr/WAM and the Subheading 4.1 to build your own models.
To use another matrix, install it in the EuGene tree and edit the wam_dataset key in the configuration file:
% cd $EUGENEDIR/models/WAM % wget http://eugene.toulouse.inra.fr/Downloads/WAM_myspeciesgroup_date.tar.gz % gzip -cd WAM_myspeciesgroup_date.tar.gz | tar xvf – % vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg wam_dataset=myspeciesgroup
3.3.2 Automatic Detection of Non-canonical Splice Sites
The introns of eukaryotic genomes start on a so-called donor site, in which the conserved dinucleotide “GT” appears. They end with a so-called acceptor site in which the conserved “AG” dinucleotide appears. These GT/AG splice sites are said to be canonical. Some species have non-canonical splice sites, in which the “GC” dinucleotide appears instead of “GT,”, for example. EuGene automatically detects the presence of non-canonical sites based on transcript alignments. A conserved dinucleotide appearing in more than 1% (the default value of parameter noncansite_required_percent) of the donor or acceptor sites detected in the alignments is activated as a non-canonical conserved dinucleotide (donor or acceptor): the EuGene graph is then edited to respectively allow the passage of the exon state to the intron state, or from intron to exon. However, this is only allowed if the alignment of a transcript confirms the exact position of the non-canonical splice site.
The detection of too many distinct non-canonical conserved dinucleotides is a warning about the quality of the alignment: poor alignment by the alignment program, poor quality transcriptome, for example. Thus, if more than 3 (the default value of parameter max_noncansite_candidate_nb) non-canonical donor or acceptor conserved dinucleotides exceed the frequency threshold noncansite_required_percent, then EuGene stops.
3.4 The “AnnotaStruct” Generic Plug-In
Additional information is sometimes available to aid in the structural annotation of a genome, for example, results of proteomic analyses which provide information on translated DNA regions or TSS (transcription start site) mapping results. EuGene, through its generic “AnnotaStruct” plug-in, can integrate various information as soon as they are formatted as a GFF3 file. The $EGNEP/ADDITIONAL directory should contain the files to integrate with “AnnotaStruct”.
In practice, to integrate a new data source, two files must be created: the raw data file in GFF3 format, and the configuration file describing how to integrate these data. The example below shows how to integrate the mapping of proteomic analysis results.
3.4.1 Expected GFF3 Format
It must be a valid GFF3 file. The features allowed in the third column are CDS , five_prime_UTR , three_prime_UTR , UTR, intron , exon, transcript_region , ncRNA, or intergenic_region. It is possible to add an Ontology_term attribute [12] in the ninth column to provide additional precision on the nature of the feature (Table 2).
The following example file describes four translated regions. The term SO:0000004 indicates that these are “interior coding exons”.
% more $EGNEP/ADDITIONAL/mygenome.MQ.peptides.gff3 Chr1 MaxQuant CDS 72033 72080 . + . ID=regionMQ.323.1;Ontology_term=SO:0000004 Chr1 MaxQuant CDS 73072 73119 . + . ID=regionMQ.325.1;Ontology_term=SO:0000004 Chr3 MaxQuant CDS 3391 3471 . + . ID=regionMQ.326.1;Ontology_term=SO:0000004 Chr3 MaxQuant CDS 8614 8664 . + . ID=regionMQ.327.1;Ontology_term=SO:0000004
3.4.2 The “AnnotaStruct” Configuration File
The AnnotaStruct configuration file describing how to integrate the information source in EuGene is composed of 24 lines. Each line is a parameter of the “AnnotaStruct” plug-in, described in the EuGene C++ program documentation available here: http://eugene.toulouse.inra.fr/Downloads/20180920/EuGeneDoc.pdf (Subheading 2.4.5.1). We provide an “AnnotaStruct” template file $EGNEP/cfg/plugin_AnnotaStruct_template.cfg in which the values are all equal to zero, so its use without any change would have no effect.
3.4.3 Example: Integrating the Mapping of Proteomic Analysis Results
Create the data file in GFF3 format according to the specifications described in Subheading 3.4.1, then copy the template configuration file and edit it:
% cp $EGNEP/cfg/plugin_AnnotaStruct_template.cfg $EGNEP/ADDITIONAL/plugin_AnnotaStruct_Proteomics.cfg % vi $EGNEP/ADDITIONAL/plugin_AnnotaStruct_Proteomics.cfg AnnotaStruct.CDS*[CPT] 2
A positive score on AnnotaStruct.CDS*[CPT] allows each position of a CDS feature to vote in the EuGene graph in favor of predicting a translated region. The higher the value, the more the information is taken into account. We recommend a value between 0.1 and 3.
Then edit the EuGene configuration file: for each additional information source, add a block of two lines. Below is an example showing how to integrate the source Z (Z must be a number):
% vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg additional_Z_file=%i/ADDITIONAL/mygenome.MQ.peptides.gff3 additional_Z_cfg_template=%i/ADDITIONAL/plugin_AnnotaStruct_Proteomics.cfg
Parameter details:
-
additional_Z_file. Absolute path of the GFF3 data file.
-
additional_Z_cfg_template. Absolute path of the corresponding “AnnotaStruct” configuration file.
Then list the numbers of the sources of information to be included in the recipe. Below is an example to integrate the source Z (Configured as described above):
% vi $EGNEP/cfg/egnep.myspecies.recipe1.cfg additional_list=Z
-
additional_list. List of the numbers of sources of information to use in the recipe. (Can be empty).
The page http://eugene.toulouse.inra.fr/Configuration provides more examples of AnnotaStruct formatted GFF3 files and configuration files.
3.5 Repeat Region Detection
The objective of the EuGene pipeline is not to annotate repeated elements. Thus the pipeline masks the repeated regions and then annotates the masked genome. In the default configuration, three tools are executed to search for repeated regions of the genome:
-
Red [13].
-
LTRHarvest [14].
-
A search for similarities (NCBI BLASTX) with a library of proteins from repeated regions. This library consists of two datasets: (a) RepBase proteins [15] and (b) proteins derived from specific repeat regions of the annotated species. The automatic procedure for detecting proteins (b) is as follows: EuGene annotates the genome the first time, extracts the predicted proteins, and searches among them for those with strong similarities with RepBase (following a BLASTP, proteins that align with a RepBase protein on more than 80% of their length are conserved). If you already have a repeat library of your species, you can avoid library automatic building (b) by specifying the path of your own dataset in the species_repeat_domains parameter.
The genomic regions detected by one of these three approaches are masked before annotation. Since there may be false positives in the masking results, it is necessary to protect the regions that are expressed. For this, the transcribed regions, that is to say having similarities with a transcriptome or a proteome, to be preserved (those whose “_ preserve” parameter is equal to 1) or those detected as known ncRNAs, are unmasked. Before anything, we ensure, as much as possible, that these unmasked transcribed/translated regions do not come from repeated regions. For instance, for this purpose the proteomes for which the value of blastx_db_Y_remove_repet is equal to 1 are cleaned, i.e. the proteins that align with a RepBase protein are deleted before searching for similarities.
3.6 Non-coding RNA Annotation
In addition to protein-coding genes, EuGene is able to annotate non-coding genes. Three mechanisms have been set up with this purpose:
-
First, EuGene integrates the results of three ncRNA predictors, tRNAScan-SE, RNAmmer, and cmsearch on Rfam, which look for ncRNAs from known families. These RNAs are identified in the GFF3 output file by additional fields in column 9 (see Subheading 3.9). It is possible to disable the use of these predictors with the options --no_ncrna_detection, --no_rfamscan, --no_trnascan, and/or --no_rnammer in the main command line $EGNEP/bin/int/egn-euk.pl.
-
A transcriptome built from total RNA libraries (e.g., without selection of mRNAs by poly(A) tail) contains sequences from mRNA and non-coding RNA. To integrate this type of data, it is possible to modify the behavior of EuGene so that when a transcript aligns in a region of low coding potential, EuGene can potentially detect a ncRNA. To enable this behavior, the parameter Est.mRNAOnly[CPT] must be equal to 0:
% vi $EGNEP/cfg/euk/plugin_Est.cfg Est.mRNAOnly[CPT] 0
-
A transcript support (of size greater than 200 nt by default) without significant coding potential (of size greater than 40 amino acids by default) is annotated by EuGene as a long ncRNA. The default values can be modified by editing the Output.MinCDSLen and Output.MinRescueTranscriptLen parameters in the $EGNEP/cfg/euk/egn_euk_generic.cfg file.
3.7 Computational Management
When running EuGene, only previously unfinished calculations are started: if the first run is not completed, the second run (using the same workingdir directory) will only run the computations that miss results. This can save a lot of time when annotating a large genome. Although EuGene has been optimized to reduce the duration of the longest tasks, some unavoidable expensive calculations of the pipeline will always require a significant execution time (e.g., search for some long Rfam ncRNAs).
Testing the existence of an output file is not enough to ensure that a calculation has been completed correctly. Thus, for each calculation, when its execution ends without error, a file bearing the name of the result file followed by the “.success” extension is created. For example, at the end of the tRNA search on chromosome Chr2 with the tRNAScan-SE software, the file /path/of/myworkingdir/0001/Chr2/Chr2.trnascan.gff3.success is created. When EuGene restarts, a process will be executed only if the corresponding “.success” file does not exist. This is useful for restarting EuGene following an interruption due to an error or when modifying a recipe.
Consider that we created a first recipe with two proteomes numbered 1 and 2 (blastx_db_list=1 2) and we now want to integrate another proteome numbered 4. One can modify the list blastx_db_list=1 2 4 in the configuration file and run again the same EuGene command: in this case, only the protein similarity calculations with the proteome 4 will be executed.
3.8 Advanced Setup
3.8.1 Changing the Minimum Size of Introns
By default EuGene will not predict introns shorter than 40 nt. If you want to change this minimum value (example to assign a minimum size of 35 nt), edit the corresponding intron.dist file and specify that intron lengths n = 35 and n + 1 = 36 are allowed. Longer intron lengths will be extrapolated.
% vi $EUGENEDIR/models/intron.dist 35 0.0 36 0.0 % cd $EUGENEDIR % make -i install
The “-i” option forces the compilation/installation even if the latex documentation compilation failed (which may happen on some systems).
3.8.2 Performing a Strand-Specific Annotation
EuGene can be run simultaneously on both strands or independently on each strand. Independent strand runs allow predicting overlapping genes in eukaryotic mode. To use this behavior, set independent_strand_annotation to 1 in the EuGene configuration file.
3.8.3 Annotating Atypical Gene Structures
The publication “Improved methods and resources for paramecium genomics: transcription units, gene annotation and gene expression” by Olivier Arnaiz et al. [16] illustrates the high flexibility of EuGene to annotate atypical gene structures. It also presents alternative ways to integrate RNA-seq data.
3.8.4 Annotating a Heterozygous Genome with Separated Haplotypes
Some genome assemblers (e.g., FALCON) can generate a primary haplotype genome sequence and an additional assembly of alternate alleles (haplotigs). To annotate such genome assembly we suggest running the Eugene pipeline twice but with the same sources of evidence: first on the primary contigs and then on the haplotigs. As only the best alignment of the transcript is kept by the pipeline, a single annotation run on the concatenation of both genomes will degrade the prediction of the unsupported allele.
3.9 Output Files
3.9.1 EuGene Output Files
Eugene generates ten files in the directory /path/of/myoutdir. The name of each file starts with the prefix defined in the output_prefix key. If the prefix is myspecies.20180801, here are the created files:
-
myspecies.20180801.gff3. Structural annotation in GFF3 format. The gene Ids are generated from the chromosome name followed by the letter “g” followed by the gene number. Genes are numbered by steps of 10 units, according to their genomic positions. To change this step, edit the “step” value in the prg_consolidate_gff3 key in the EuGene configuration file. If the chr_sorting_by_size value is equal to 1, the genomic sequences are sorted by decreasing size before the annotation, so the gene number 1 is the first annotated gene on the largest sequence. The genes encoding proteins are described using the following features: gene, mRNA, exon, three_prime_UTR, five_prime_UTR , and CDS. The ncRNAs are described with the following features: gene, rRNA, tRNA , and ncRNA. The column nine contains additional attributes describing the features (see Table 3).
-
myspecies.20180801. Multi-FASTA file of the genomic sequences, optionally sorted by decreasing size if the chr_sorting_by_size value is equal to 1.
-
myspecies.20180801.*.fna. Five multi-FASTA files corresponding to CDSs, genes, mRNAs, ncRNAs, and protein sequences.
-
myspecies.20180801.general_statistics.xls. Statistics on the annotation including, among others, the number of predicted genes, the protein-coding and non-coding genes, the average lengths of various features, and the average GC% of ncRNA genes. This can give a first feedback on the annotation quality.
-
myspecies.20180801.statistics_per_gene.xls. Tabulated file that gives per-mRNA and ncRNA statistics such as length, number of introns, or 5′ UTR region length.
-
myspecies.20180801.egnep_report.txt. Text file containing information on the different stages of the pipeline. The first part is dedicated to the alignment of transcriptomes. For each transcriptome, the number of raw sequences, and the number and the percentage of aligned sequences remaining after the various filters (which correspond to the sequences actually used for the annotation) are reported. If this latter value is low, it may be important to understand why: is the assembly of the transcriptome of poor quality? Is there a contamination? Are the mapping parameters suitable? The second part lists the detected non-canonical splice sites (see Subheading 3.3.2). Finally, the third section gives the percentage of the genome that has been considered as a repeated region, both before and after the unmasking caused by aligned transcripts, ncRNA, or proteins (see Subheading 3.5).
3.9.2 Upload Annotation Files in a Genome Browser
To facilitate the visualization of the genome annotation produced, the program convert_egn2mygenomebrowser.pl can generate all the files needed to build an instance of a genome browser such as JBrowse [17]. The generated files can be used directly in MyGenomeBrowser [18]. From the outdir and workingdir directories, and the EuGene configuration file, the program puts together the genome, the annotation results, and all the evidences used for the annotation in a tarball named MyGenomeBrowser.tar.gz. Example:
% $EGNEP/bin/int/misc/convert_egn2mygenomebrowser.pl \ --egnep_outdir /path/of/myoutdir \ --egnep_workdir /path/of/myworkingdir \ --cfg /path/of/myeugeneconffile.cfg
3.10 Prokaryotic EuGene Version
This section describes the main differences between the eukaryotic version of the pipeline described in the previous sections and the prokaryotic version presented in detail in [8, 19].
-
The topology of the prokaryotic graph that describes the genome structure is different: it does not allow for introns but instead enables overlapping CDS (on the same or on opposite strands). The prediction of UIRs (Untranslated Internal Regions: regions transcribed between two genes) can gather several genes into a single transcription unit. These operons are annotated in the GFF3 output file.
-
EuGene is run independently on each strand, allowing, for example, the detection of antisense ncRNAs.
-
An additional plug-in that predicts potential transcription starts (TSS) is used. This prediction is based on the detection of abrupt changes in the level of expression of RNA-seq data aligned with the genome.
-
Prokaryotic genomes being compact and containing little repeated DNAs, there is no repeat masking process.
-
It is possible to integrate expression data in different formats: oriented single-end or oriented paired-end reads in FASTA or FASTQ format; mapped reads in Bam/Sam, Bed, or Wig format, as well as tiling array data as a pair of ndf/pair files.
In practice:
To install and configure the prokaryotic pipeline, download and untar the http://eugene.toulouse.inra.fr/Downloads/egnpp-Linux-x86_64.1.3.tar.gz tarball and follow the instructions in the QUICKSTART file. The EGNPP environment variable must be set. The command to run is:
% $EGNPP/bin/int/egn-prok.pl \ --indir /path/of/myindir \ --outdir /path/of/myoutdir \ --cfg /path/of/myeugeneppconffile.cfg
The indir directory must contain two directories named genome and data. The genome directory must contain the genomic sequences to annotate and the data directory all the evidences used in the annotation recipe.
In the configuration file:
-
The reference proteome must be specified in the training_db_file parameter.
-
The expression data to be used in the recipe are described using evidence_* keys. For each type of data, add a block of 3 to 5 lines. Below is an example in which one integrates oriented paired-end RNA-seq data in FASTQ format, termed A:
evidence_A_pat=*.fastq.gz evidence_A_format=fastq evidence_A_type=ope evidence_A_oriented_end=1 evidence_A_small=0 evidence_list=A
Parameter details:
-
evidence_A_pat. Pattern used inside the /path/of/myindir/data/ directory to list all the data files to process.
-
evidence_A_format (possible values: fastq, fasta, wig, sam, bam, pair). File format.
-
evidence_A_type (possible values: ose, ope). Respectively, “oriented single-end” or “oriented paired-end.” The key is compulsory if the files are in FASTA or FASTQ format.
-
evidence_A_oriented_end (possible values: 1, 2). Number of the read end that gives the orientation (It depends on the extraction and sequencing protocols).
-
evidence_A_small (possible values: 0, 1). A value of 1 specifies that the RNA-seq is small RNAs library.
Note: to integrate Roche NimbleGen tiling array hybridization results, three parameters need to be specified: evidence_A_pat, the pattern allowing to list the files in NimbleGen PAIR format, evidence_A_format with a value set to “pair,” and evidence_A_ndf, the absolute path of the NimbleGen-formatted design (ndf) file.
3.11 Limitations
The current eukaryotic gene model does not allow for the prediction of ncRNAs or other genes inside the introns of genes. When such intronic genes exist, they tend to generate the prediction of split genes so that the intronic gene can be predicted.
The EuGene C++ software contains plug-ins capable of handling the presence of frameshifts in CDS and detecting splice variants based on transcript alignments [20]. These plug-ins are not enabled in the standard version of the pipeline because the goal is to predict a full genome reference annotation.
The EuGene pipeline is not suitable for the annotation of chloroplasts and mitochondria. In these cases, it is best to use specialized tools.
4 Companion Tools
4.1 Building New Splice Sites WAM Matrices for a New Species or Group of Species
EuGene uses statistical models (Weight Array Method matrices) that have already been trained for the detection of splice sites. The available models are described here: http://eugene.toulouse.inra.fr/WAM /.
If no available model matches your species, you can build a new WAM matrix. The prerequisite is to have several complete genomes of closely related species, and for each of them a good quality transcriptome, as complete as possible. Indeed, to train the models, the transcripts must include a sufficiently large number of genes of various families to be able to capture the variability of the signals around the splice sites.
In practice:
Copy the template configuration file and edit it to fit your data:
% cp $EGNEP/cfg/euk/egn_build_wam.cfg $EGNEP/cfg/egn_build_wam_myspeciesgroup.cfg
Specify the name of the group of species and the genome/transcriptome pairs:
% vi $EGNEP/cfg/egn_build_wam_myspeciesgroup.cfg wam_species=myspeciesgroup wam_dataset_list=E F wam_dataset_E_genome=/path/of/genomeE wam_dataset_E_transcriptome=/path/of/transcriptomeE wam_dataset_F_genome=/path/of/genomeF wam_dataset_F_transcriptome=/path/of/transcriptomeF
Then run:
% $EGNEP/bin/int/egn_build_wam.pl --cfg $EGNEP/cfg/egn_build_wam_myspeciesgroup.cfg\ --outdir /path/of/wamoutdir
At the end of the execution, a tarball /path/of/wamoutdir/myspeciesgroup.tar.gz containing the donor and acceptor WAM matrices is created. See instructions in Subheading 3.3.1 to use these models.
More information about the program options can be obtained with the command:
% $EGNEP/bin/int/egn_build_wam.pl --help
4.2 Transferring Annotations Between Genome Releases
The EuGene tarball includes the program $EGNEP/bin/int/misc/egn_annotation_transfer.pl which allows transferring the structural annotation of a sequence to another sequence. There is no gene discovery, only the genes of the initial annotation are transferred to the new sequence. The genes annotated on the new sequence will have a “valid” structure, one that is compatible with EuGene’s internal gene model (e.g., every CDS starts with a start codon, ends with a stop codon, and has a length which is a multiple of 3). The name of the source gene is indicated in the “Alias” attribute of the output GFF3 file. The program is useful to transfer the annotation between genotypes/strains of the same species while preserving the correspondence between genes.
The pipeline is detailed here: http://eugene.toulouse.inra.fr/Tools/egn_annotation_transfer.html
Usage:
% $EGNEP/bin/int/misc/egn_annotation_transfer.pl --help --scaffoldsgenomic sequences to annotate (multi-fasta) --ref_scaffolds reference genomic sequences (multi-fasta) --ref_gff3 reference structural annotation (gff3) --outfile transferred structural annotation (gff3) --workingdir working directory --cfg configuration file
To contact the authors, use eugene-help@groupes.renater.fr. To receive information and updates on EuGene, subscribe to eugene-info mailing list https://groupes.renater.fr/sympa/info/eugene-info.
References
Foissac S, Bardou P, Moisan A, Cros MJ, Schiex T (2003) EUGENE'HOM: a generic similarity-based gene finder using multiple homologous sequences. Nucleic Acids Res 31(13):3742–3745
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964
Lagesen K, Hallin PF, Rødland E, Stærfeldt HH, Rognes T, Ussery DW (2007) RNammer: consistent annotation of rRNA genes in genomic sequences. Nucleic Acids Res 35(9):3100–3108
Nawrocki EP, Eddy SR (2013) Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29:2933–2935
Schiex T, Moisan A, Rouzé P (2001) Eugène: an eukaryotic gene finder that combines several sources of evidence. In: Gascuel O, Sagot MF (eds) Computational biology. JOBIM 2000. Lecture notes in computer science, vol 2066. Springer, Heidelberg
Foissac S, Gouzy J, Rombauts S, Mathé C, Amselem J, Sterck L, Van de Peer Y, Rouzé P, Schiex T (2008) Genome annotation in plants and fungi: EuGene as a model platform. Curr Bioinforma 3(2):87–97
Bellman R (1957) Dynamic programming. Princeton Univ. Press, Princeton, NJ
Sallet E, Roux B, Sauviac L, Jardinaud MF, Carrere S, Faraut T, de Carvalho-Niebel F, Gouzy J, Gamas P, Capela D, Bruand C (2013) Next-generation annotation of prokaryotic genomes with EuGene-P: application to Sinorhizobium meliloti 2011. DNA Res 20(4):339–354
Lafferty J, McCallum A, Pereira FCN (2001) Conditional random fields: probabilistic models for segmenting and labeling sequence data. ICML '01 proceedings of the eighteenth international conference on machine learning
Badouin H et al (2017) The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546(7656):148–152
Zhang MQ, Marr TG (1993) A weight array method for splicing signal analysis. Bioinformatics 9(5):499–509
Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M (2005) The sequence ontology: a tool for the unification of genome. Genome Biol 6:R44
Girgis HZ (2015) Red: an intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinformatics 16:227
Ellinghaus D, Kurtz S, Willhoeft U (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9:18
Bao W, Kojima KK, Kohany O (2015) Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6:11
Arnaiz O, Van Dijk E, Bétermier M, Lhuillier-Akakpo M, de Vanssay A, Duharcourt S, Sallet E, Gouzy J, Sperling L (2017) Improved methods and resources for paramecium genomics: transcription units, gene annotation and gene expression. BMC Genomics 18(1):483
Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH (2009) JBrowse: a next-generation genome browser. Genome Res 19:1630–1638
Carrere S, Gouzy J (2017) myGenomeBrowser: building and sharing your own genome browser. Bioinformatics 33(8):1255–1257
Sallet E, Gouzy J, Schiex T (2014) EuGene-PP: a next-generation automated annotation pipeline for prokaryotic genomes. Bioinformatics 30(18):2659–2661
Foissac S, Schiex T (2005) Integrating alternative splicing detection into gene prediction. BMC Bioinformatics 6:25
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

{kind=link}
Cite this protocol
Sallet, E., Gouzy, J., Schiex, T. (2019). EuGene: An Automated Integrative Gene Finder for Eukaryotes and Prokaryotes. In: Kollmar, M. (eds) Gene Prediction. Methods in Molecular Biology, vol 1962. Humana, New York, NY. https://doi.org/10.1007/978-1-4939-9173-0_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-9173-0_6
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-4939-9172-3
Online ISBN: 978-1-4939-9173-0
eBook Packages: Springer Protocols