










La secuencia del ácido desoxirribonucleico (ADN) experimenta cambios continuamente, la mayoría de los cuales, los polimorfismos, no producen alteraciones en el fenotipo. Sin embargo, otros alteran la estructura de un gen, de modo que afectan a la función de la proteína que sintetizan, y dan lugar a distintas enfermedades. Estos cambios patológicos del ADN se denominan mutaciones1. La mayoría de enfermedades monogénicas son el resultado de mutaciones puntuales, por deleción, inserción o sustitución de un nucleótido por otro. Estas alteraciones se pueden clasificar en: a) silenciosas (no cambia el aminoácido); b) de cambio de sentido o missense (cambia el aminoácido); c) sin sentido o nonsense (cambia por un codón de stop), y d) de splicing, que afectan al ácido ribonucleico (ARN). También pueden encontrarse grandes deleciones (distrofia muscular de Duchenne), inserciones (hemofilia A) o expansión anormal de tripletes repetitivos (síndrome X frágil [SXF]).
Para identificar las mutaciones del ADN que causan enfermedades genéticas, hoy día hay numerosas técnicas de diagnóstico molecular. En la tabla 1 se incluyen las más utilizadas.
Puntos clave

Selección de las técnicas de diagnóstico molecular más utilizadas para identificar mutaciones causantes de enfermedades genéticas
| Amplificación enzimática del ADN PCR RT-PCR |
| Estudio de marcadores genéticos RFLP STR VNTR |
| Estudios genéticos indirectos Análisis de ligamiento SNP |
| Estudios genéticos directos Southern blot SSCP HA DGGE DHPLC MLPA FISH STS Secuenciación Arrays |
ADN: ácido desoxirribonucleico; DGGE: electroforesis desnaturalizante en gel de gradiente; DHPLC: cromatografía líquida desnaturalizante de alto rendimiento; FISH: hibridación in situ con fluorescencia; HA: análisis de heterodúplex; MLPA: amplificación con sonda múltiple dependiente de ligamiento; PCR: reacción en cadena de la polimerasa; RFLP: fragmentos de restricción de longitud polimórfica; RT-PCR: PCR en tiempo real; SSCP: polimorfismos por conformación de cadena única; SNP: polimorfismo de nucleótido único; STR: fragmentos repetitivos pequeños en tándem; STS: lugar de secuencia marcado; VNTR: fragmentos repetitivos de tamaño variable en tándem. Las abreviaturas proceden del nombre de la técnica en inglés. Véanse los epígrafes del texto para ver el nombre completo original en inglés.
En la cadena del ADN humano, en cada 100 pares de bases, aproximadamente, hay una diferencia en la secuencia de un individuo a otro. Estas diferencias se denominan polimorfismos. Como resultado, la secuencia de reconocimiento de una enzima de restricción puede estar presente en un cromosoma, pero no en otro. Los fragmentos de restricción de longitud polimórfica (RFLP, del inglés restriction fragment length polymorphisms) son variaciones del ADN en los sitios de restricción, posición en la que varían los tamaños de los fragmentos. Los RFLP no se relacionan con la mutación, pero se pueden utilizar como marcadores. Algunos RFLP se basan en la inserción o deleción de un segmento variable de ADN, y no en la pérdida o ganancia de un sitio de reconocimiento para las enzimas de restricción. Por ejemplo, una clase especial de polimorfismos resulta de la inserción, en tándem, de múltiples copias de una secuencia de ADN, conocidos como minisatélites. Esta clase de RFLP se denomina polimorfismo de repetición en tándem de número variable (VNTR, del inglés variable number tándem repeats). Los microsatélites son extensiones de ADN compuestas por unidades repetidas de 2, 3 o 4 nucleótidos, y son más frecuentes que los minisatélites2.
Lectura rápida
La mayoría de las enfermedades monogénicas son el resultado de mutaciones puntuales, por deleción, inserción o sustitución de un nucleótido por otro; estas alteraciones se pueden clasificar en: a) silenciosas (no cambia el aminoácido); b) de cambio de sentido o missense (cambia el aminoácido); c) sin sentido o nonsense (cambia por un codón de stop), y d) de splicing, que afectan al ácido ribonucleico (ARN).
El análisis de ligamiento (linkage analysis) es una técnica designada para detectar la segregación de 2 o más loci génicos muy cercanos, localizados en el mismo cromosoma. se utiliza para identificar el denominado "haplotipo de riesgo" asociado a una enfermedad3. Al ser un método de diagnóstico indirecto, en los estudios familiares es necesario estudiar al mayor número de miembros posible, especialmente a los individuos sanos y afectados, en varias generaciones. Es una técnica muy útil en el diagnóstico prenatal. Con los estudios de ligamiento no se identifican las mutaciones de un gen, ya que es un método indirecto de análisis del ADN, cuyos resultados se basan en métodos estadísticos sofisticados con soporte de software para su aplicación.
Southern blotEn 1975, Ed Southern desarrolló una técnica que permitía el estudio de fragmentos de ADN cortados mediante enzimas de restricción a partir de ADN purificado. Las enzimas (endonucleasas) de restricción cortan el ADN en lugares específicos (secuencia de reconocimiento), y lo fragmentan en miles de segmentos de múltiples tamaños que, posteriormente, se ordenan por tamaño con una electroforesis en gel de agarosa. Los fragmentos contenidos en el gel se transfieren a una membrana de nitrocelulosa o de nailon para su desnaturalización (separación de las 2 hebras de ADN) con un álcali y fijación mediante un ligamiento cruzado con luz ultravioleta. El ADN del individuo, fragmentado y fijado, se incuba con un pequeño fragmento conocido (sonda) de ADN (genómico o complementario [ADNc]) complementario al segmento del ADN de interés para el estudio. La sonda, que previamente se ha marcado, producirá una señal visible si hibrida con el fragmento complementario del ADN problema. Cuando se analiza el ARN, la técnica se denomina Northern blot4.
Reacción en cadena de la polimerasaLa reacción en cadena de la polimerasa (PCR, del inglés polimerase chain reaction) es una técnica que permite generar cantidades ilimitadas de una determinada secuencia de ADN. La PCR puede amplificar selectivamente una sola molécula de ADN o ARN varios millones de veces en pocas horas. La técnica consiste en una amplificación enzimática de un fragmento de ADN localizado y limitado entre 2 oligonucleótidos iniciadores ("cebadores" o primers), cada uno complementario de las secuencias situadas a ambos extremos del fragmento diana. La reacción da lugar a 2 cadenas nuevas de ADN complementarias, que forman una segunda copia del fragmento original. La reacción se repite 25 a 30 ciclos, cada uno de los cuales consta de 3 reacciones controladas en tiempo y temperatura, que se llevan a cabo en termocicladores automáticos. Los 3 pasos en cada ciclo son: a) desnaturalización del ADN de cadena doble (93–95ºC); b) hibridación del cebador al ADN (50–70ºC), y c) síntesis del ADN, utilizando una polimerasa de ADN termorresistente (Taq) (70–75ºC). La PCR es una técnica rápida y barata, que requiere menos cantidad de muestra del paciente que cualquier otro método de análisis de ácidos nucleicos. Es además muy sensible, lo que la hace muy susceptible a la contaminación5.
Reacción en cadena de la polimerasa en tiempo realUna secuencia de aminoácidos de un polipéptido parcialmente conocida puede utilizarse para obtener la información de la secuencia necesaria para una PCR. A partir de su ARN mensajero (ARNm) uno puede obtener ADNc y determinar la secuencia de la cadena de sentido y antisentido para preparar oligonucleótidos cebadores apropiados. El ADNc se obtiene de la transcripción inversa del ARNm. La PCR de una transcripción inversa (PCR en tiempo real [RT-PCR, del inglés real-time PCR]) puede utilizarse cuando las secuencias de exones conocidas están ampliamente separadas dentro de un gen3.
Lectura rápida
Para la identificación de las mutaciones del ADN que causan enfermedades genéticas, hoy día hay numerosas técnicas de diagnóstico molecular. Las más utilizadas son el Southern blot, la reacción en cadena de la polimerasa (PCR), la secuenciación y la amplificación múltiple con sonda dependiente de ligamiento (MLPA).
Southern blot Es una técnica diseñada para detectar la segregación de 2 o más loci génicos muy cercanos, localizados en el mismo cromosoma. Se utiliza para identificar el denominado "haplotipo de riesgo" asociado a una enfermedad. Al ser un método de diagnóstico indirecto, en los estudios familiares es necesario estudiar al mayor número de miembros posible, especialmente a los individuos sanos y afectados, en varias generaciones. Es una técnica muy útil en el diagnóstico prenatal.
Reacción en cadena de la polimerasa. La PCR es una técnica que permite generar cantidades ilimitadas de una determinada secuencia de ADN. La PCR puede amplificar selectivamente una sola molécula de ácido desoxirribonucleico (ADN) o ARN varios millones de veces en pocas horas.
Una hebra de la doble cadena de ADN se pliega de una forma específica cuando se introduce en una solución no desnaturalizante. Un cambio en la secuencia de ADN produce cambios en la estructura de plegamiento, que puede alterar la movilidad de la conformación en un gel de agarosa no desnaturalizante. La sensibilidad de esta técnica se encuentra entre el 35 y el 100%, aunque en la mayoría de los estudios se ha detectado más del 80% de las mutaciones. La mayor limitación del polimorfismo por conformación de cadena única (SSCP, del inglés single-strand conformation polymorphism) es el tamaño del fragmento, en el que se ha demostrado que la sensibilidad varía dependiendo del tamaño del fragmento3,6.
Análisis de heterodúplexEn una electroforesis en geles no desnaturalizantes, los heterodúplex tienen una movilidad más lenta que los homodúplex. Inicialmente, esta técnica se describió para detectar inserciones y deleciones, pero finalmente también era útil para detectar mutaciones de una base. Esta técnica se emplea para fragmentos superiores a 1kb de tamaño, y se ha observado que la eficacia para detectar mutaciones disminuye en fragmentos más grandes7.
Electroforesis desnaturalizante en gel de gradienteEsta técnica consiste en que la doble cadena de ADN atraviese un gel, en el cual la concentración desnaturalizante va en aumento. Como consecuencia, la doble hebra se desnaturalizará parcialmente y retardará la movilidad electroforética. La sustitución de un nucleótido alterará la unión de ambas cadenas y, por lo tanto, la movilidad en el gel. La sensibilidad de esta técnica es del 95-100% para fragmentos por encima de 500 pb8.
Cromatografía líquida desnaturalizante de alto rendimientoEl objetivo de esta técnica es detectar mutaciones a partir de las diferentes propiedades de unión de los homodúplex y heterodúplex. Se produce una retención diferente de las moléculas en la columna de cromatografía en condiciones de desnaturalización térmica parcial. Actualmente, la cromatografía líquida desnaturalizante de alto rendimiento (DHPLC, del inglés denaturing high-performance liquid chromatography) tiene aplicaciones tanto en la investigación como en el diagnóstico. Presenta una sensibilidad elevada, del 91-100%. Las ventajas de esta técnica son su alta sensibilidad y la alta capacidad de procesamiento, y las desventajas son el coste elevado del equipamiento necesario y la necesidad de conocer la temperatura precisa para el análisis de cada fragmento5,9.
SecuenciaciónEl conocimiento de la secuencia completa de nucleótidos de un gen ofrece información importante acerca de su estructura, función y relación evolutiva con otros genes similares. Por ello, el desarrollo de métodos relativamente simples de secuenciación del ADN, en la década de los setenta, tuvo un gran impacto en la genética, inicialmente en la investigación básica y, posteriormente, en su aplicación como técnica de diagnóstico molecular. Los métodos básicos de secuenciación del ADN son 2: un método de desdoblamiento químico (Maxam, 1977) y un método enzimático (Sanger, 1981)1,5,10.
La secuenciación por degradación química se basa en el desdoblamiento específico del ADN por medio de ciertas sustancias químicas. En cada reacción, se produce un juego de fragmentos de ADN de longitudes diferentes. Las 4 mezclas de las reacciones, una para cada una de las bases, se corren en calles separadas de una electroforesis en gel de poliacrilamida. Cada una de las 4 calles representa a una de las 4 bases G, A, T o C. La secuencia se lee en dirección opuesta a la migración para conseguir una lectura de 5' a 3'.
La secuenciación por terminación de la cadena se utiliza mucho más y se basa en el principio de que la síntesis del ADN se termina cuando, en lugar de un desoxinucleótido (dNTP) normal, se utiliza un didesoxinucleótido (ddNTP) que es un análogo de un dNTP normal, pero que no presenta un grupo hidroxilo en la posición del carbono 3'. La síntesis del ADN se inicia con un cebador y uno de los 4 ddNTP marcado con 32P en el grupo fosfato. Esto se realizará para las 4 bases, las 4 reacciones por separado darán lugar a un juego de fragmentos de tamaños definidos, de acuerdo con las posiciones de los nucleótidos, donde la síntesis del nuevo ADN se ha terminado. Estos fragmentos se separan de acuerdo con el tamaño mediante una electroforesis en gel de poliacrilamida, al igual que el método anterior. Esto permitirá determinar la secuencia nucleotídica en dirección 5' a 3'4.
La secuenciación del ADN a gran escala requiere procedimientos automatizados, basados en la marcación del ADN con fluorescencia, y sistemas de detección adecuados. La secuencia se lee y registra en formato electrónico y se visualiza como picos alternantes de uno de los 4 colores, lo cual representa la alternancia de los nucleótidos en su posición secuencial.
Lectura rápida
Secuenciación. La técnica de secuenciación puede realizarse por 2 procedimientos. La secuenciación por degradación química se basa en el desdoblamiento específico del ADN por medio de ciertas sustancias químicas. En cada reacción se produce un juego de fragmentos de ADN de longitudes diferentes. La secuenciación por terminación de la cadena se utiliza mucho más y se basa en el principio de que la síntesis del ADN se termina cuando, en lugar de un desoxinucleótido (dNTP) normal, se utiliza un didesoxinucleótido (ddNTP), que es un análogo de un dNTP normal, pero que no presenta un grupo hidroxilo en la posición del carbono 3'. La síntesis del ADN se inicia con un cebador y uno de los 4 ddNTP marcado en el grupo fosfato. Esto se realizará para cada una de las 4 bases, las 4 reacciones por separado darán lugar a un juego de fragmentos de tamaños definidos, de acuerdo con las posiciones de los nucleótidos, en donde la síntesis del nuevo ADN se ha terminado. Estos fragmentos se separan de acuerdo con el tamaño, mediante una electroforesis en gel de poliacrilamida.
Amplificación múltiple con sonda dependiente de ligamiento. La técnica de amplificación con sonda múltiple dependiente de ligamento (MLPA) permite la detección de deleciones y duplicaciones del genoma, así como la detección de anomalías cromosómicas (trisomías). La MLPA consiste en el análisis de determinadas regiones del ADN, mediante el estudio de 40 sondas que hibridan en diferentes puntos de la región del genoma que se quiere estudiar. Estas sondas se ligan y se amplifican utilizando un mismo par de cebadores. Los fragmentos resultantes de la hibridación se visualizarán en un gel, y si se aprovecha la diferencia de tamaño de cada sonda, se podrá identificar pérdidas o ganancias de material genómico, teniendo siempre como referencia un control sano.
Es una técnica que permite detectar deleciones y duplicaciones en distintos genes, así como trisomías. La amplificación con sonda múltiple dependiente de ligamiento (MLPA, del inglés multiplex ligation-dependent probe amplification) consiste en el cribado de determinadas regiones del ADN mediante el estudio de 40 sondas que hibridarán en diferentes puntos de la región que se quiere estudiar, se ligarán y se amplificarán utilizando un mismo par de cebadores. Los fragmentos se visualizarán en un gel, y aprovechando la diferencia de tamaño de cada sonda, se podrán identificar pérdidas o ganancias de material genético, teniendo siempre como referencia un control sano. Esta técnica es relativamente sencilla, barata y bastante sensible. La desventaja es que sólo es útil para mutaciones puntuales conocidas, no sirve para cribado de mutaciones no conocidas11. En la figura 1 se puede ver la resolución de la MLPA en relación con otras técnicas de diagnóstico molecular.
 con otras técnicas. La MLPA puede detectar un rango de alteraciones genómicas más amplio que el resto, desde mutaciones puntuales hasta grandes deleciones, además de duplicaciones cromosómicas. Tomada de www.mlpa.com (2007). CGH: Hibridación genómica comparada; FISH: hibridación in situ con fluorescencia; M-FISH: técnica de múltiple FISH o hibridación in situ con fluorescencia; PTT: test de proteíneas truncadas; SKY: cariotipado espectral; SNP: polimorfismo de nucleótido único; SSSCP: polimorfismo por conformación de cadena única; STS: lugar de secuencia marcado.")
Comparación de la multiplex ligationdependent probe amplification (MLPA) con otras técnicas. La MLPA puede detectar un rango de alteraciones genómicas más amplio que el resto, desde mutaciones puntuales hasta grandes deleciones, además de duplicaciones cromosómicas. Tomada de www.mlpa.com (2007). CGH: Hibridación genómica comparada; FISH: hibridación in situ con fluorescencia; M-FISH: técnica de múltiple FISH o hibridación in situ con fluorescencia; PTT: test de proteíneas truncadas; SKY: cariotipado espectral; SNP: polimorfismo de nucleótido único; SSSCP: polimorfismo por conformación de cadena única; STS: lugar de secuencia marcado.
Actualmente hay kits comerciales para el diagnóstico de distintos síndromes y enfermedades genéticas, como el síndrome de Prader-Willi, el síndrome de Angelman, la enfermedad de Alzheimer, el síndrome de Charcot-Marie-Tooth, el síndrome de Cornelia de Lange (sCdL), el síndrome de Turner, el síndrome de Rett, el estudio de regiones cromosómicas relacionadas con retraso mental, diferentes tipos de cáncer o estudios de ADN mitocondrial.
MicroarraysEsta nueva técnica se basa en la colocación ordenada sobre un sustrato —array— (plástico, cristal, membrana) de secuencias cortas de nucleótidos o ADN de doble cadena, diseñadas a partir de ADNc (sin intrones). El material genético que se coloca en cada array es conocido y cada celda representa un gen. El primer paso es marcar el material (ADN) que se va a estudiar y, a continuación, se pone en contacto con el ADN del array. Se producirá hibridación en las celdas en las que haya una complementariedad entre las sonda que contiene y el ADN problema. La visualización de esta hibridación se realizará a través de microscopia conofocal. El grado de expresión de un gen se refleja en el número de copias de ARNm presentes en la muestra-problema, que es proporcional al nivel de señal detectado. Los resultados obtenidos se analizan con herramientas bioinformáticas.
La aplicación más conocida de los arrays de ADN es la determinación de perfiles de transcripción, pero también es útil en genómica estructural. Los arrays de hibridación genómica comparada (CGH-array) es una técnica que combina la tecnología de los microarrays con la (CGH) donde la hibridación se realiza en segmentos de ADN de tamaño menor clonados en forma de cromosomas artificiales bacterianos. Esta técnica permite detectar variaciones en la dosificación o el número de copias del genoma con un nivel alto de resolución. Es una metodología muy empleada en el diagnóstico molecular de tumores12,13.
Ejemplos seleccionados de síndromes pediátricos dismórficos susceptibles de diagnóstico molecularSíndrome X frágilEl SXF (OMIM # 300624) es la causa más común de retraso mental hereditario, y afecta a uno de cada 4.000 varones y una de cada 6.000 mujeres. Se hereda de forma dominante y ligado al cromosoma X, con una penetrancia incompleta, en el 80% en varones y el 30% en mujeres. La expresividad clínica es muy variable, aunque en la mayoría de los varones afectados las manifestaciones clínicas son similares14. En el SXF hay individuos sanos que pueden transmitir el síndrome, especialmente las mujeres, ya que se estima que 1 de cada 300 mujeres de la población general es portadora. El varón también puede ser transmisor (NTM, del inglés normal transmitting male).
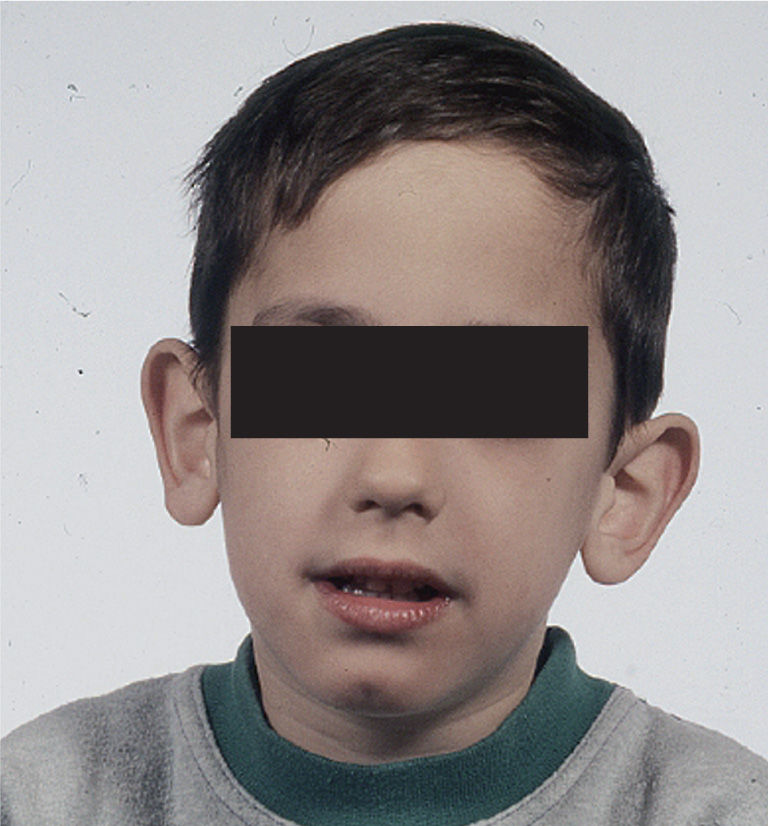
ClínicaLos hallazgos clínicos más habituales del varón afectado son: retraso mental leve-moderado, cara alargada, orejas grandes y prominentes, y macroorquidismo (testículos grandes) al llegar a la pubertad (fig. 2). En el niño pequeño, las manifestaciones clínicas pueden no ser tan evidentes y destacan el retraso en el inicio del lenguaje, la hiperactividad y/o falta de atención, y una hiperlaxitud articular15.

Varón de 5 años con síndrome X frágil. El fenotipo es característico, con cara alargada, orejas grandes y salientes y mentón prominente. Además, presentaba un retraso importante del lenguaje. El paciente tenía un expansión de ≈ 700 CGG en el gen FMR1. CGG: citosina, guanina y guanina.
En cuanto a las mujeres, no puede hablarse de ningún rasgo físico característico, y sólo un pequeño porcentaje manifiesta un retraso mental leve con dificultades de aprendizaje para las matemáticas.
Diagnóstico molecularEl análisis de la secuencia repetitiva (CGG)n del gen FMR1, localizado en la región Xq27.3, permite el diagnóstico molecular directo del SXF. Mediante la técnica del Southern blot podemos analizar el tamaño de la expansión (CGG)n y el estado de metilación de la isla CpG de FMR1, permitiendo identificar las diferentes clases de individuos: normales (5–50 CGG), premutados/as (50–200 CGG) y mutados completos (> 200 CGG)16.
También se utiliza la PCR, que no sirve para definir el estado de metilación del gen. No obstante, el análisis mediante PCR tiene importancia para el asesoramiento genético, especialmente para las madres portadoras, ya que hay una relación directa entre el número de repeticiones y la probabilidad de que se produzca el paso de premutación a mutación completa en la generación siguiente. Así, para una mujer con una premutación por encima de 90 CGG, la penetrancia es del 100%, es decir, siempre que pase el alelo mutado, éste se expandirá a mutación completa, mientras que si la mujer portadora tiene entre 50 y 60 repeticiones, la penetrancia es sólo del 18%, es decir, tan sólo en un 18% de los casos se producirá expansión16. La técnica de la PCR presenta algunas limitaciones, la principal de las cuales es que los alelos con un elevado número de repeticiones (CGG)n son muy difíciles de amplificar y, por lo tanto, las mujeres con mutación completa podrían interpretarse como falsos negativos. Para evitar riesgos, se realizan ambas, especialmente en casos de diagnóstico prenatal.
Todas las madres de los varones afectados son portadoras obligadas de la premutación, o de la mutación completa, ya que hasta la fecha no se ha descrito ninguna mutación completa de novo17.
Lectura rápida
El síndrome X frágil es la causa más frecuente de retraso mental hereditario y se diagnostica analizando la expansión del trinucleótido CGG en el gen FMR1.
El síndrome de Cornelia de Lange (SCdL) se caracteriza por retraso de crecimiento y mental, facies peculiar y oligodactilia. El 70% de los pacientes tiene mutaciones en los genes NIPBL (50%) y SMC1A (20%).
La acondroplasia es la displasia ósea más frecuente. Se hereda de forma autosómica dominante y el 98% de los afectados tienen la misma mutación en el gen FGFR3, localizado en el cromosoma 4.
El Síndrome de Cornelia de Lange (OMIM # 122470) es un trastorno del desarrollo hereditario con transmisión autosómica dominante que se caracteriza por fenotipo craneofacial distintivo con microcefalia, sinofridia y philtrum alargado con labio superior fino, anomalías en extremidades superiores (oligodactilia), disfunción grastroesofágica, cardiopatía congénita, anomalías oftalmológicas y retraso de crecimiento y psicomotor (fig. 3). Clínicamente, se distinguen 3 fenotipos: a) clásico; b) leve, y c) fenocopias o variantes. En 1933, la Dra. Cornelia de Lange lo describió por primera vez en 2 niñas. La prevalencia es variable según los estudios publicados, y oscila entre 1/10.000 nacimientos en Estados Unidos y 0,6/100.000 en Dinamarca. En 1998, tras un estudio multicéntrico, la prevalencia en España se estimó en aproximadamente 1/100.000 nacimientos.
 clásico. Obsérvese la facies típica con sinofridia y la oligodactilia bilateral.")
Aunque la mayoría de los pacientes publicados son esporádicos, una revisión de los casos familiares conocidos hasta el año 2000 concluyó que el patrón autosómico dominante era el modo de transmisión más probable, siendo en estos casos una mutación espontánea la causa del síndrome. En algunas instancias, se han descrito casos de SCdL en 2 o más individuos hijos de padres consanguíneos o en gemelos monocigóticos18.
Diagnóstico molecularAunque el SCdL es uno de los síndromes genéticos dismórficos más característicos, conocido desde hace más de 70 años, su etiología genética ha permanecido oculta hasta el año 2004. En el que un grupo de investigadores identificaron un gen candidato en la región cromosómica 5p13 gracias al estudio cromosómico de un paciente con fenotipo SCdL y portador de una traslocación cromosómica balanceada de novo t(5;13)(p13.1;q12.1). El nuevo gen se denominó (NIPBL) (nipped-B like) y en él se encontraron las primeras mutaciones en pacientes con SCdL, con lo que se concluyó que ésta era la causa del síndrome18.
Las mutaciones del gen NIPBL se encontraron tanto en los casos de SCdL clásico como leve y tanto en casos familiares como en pacientes aislados. Las mutaciones encontradas en los casos esporádicos eran deleciones e inserciones que alteraban el marco de lectura, truncando la síntesis de la proteína. En cambio, en los casos familiares, predominaban las mutaciones de cambio de sentido (missense) y punto de corte, presentes en todos los descendientes afectados, pero ausentes en los progenitores, lo que indicaba la existencia de un mosaicismo germinal18. El gen NIPBL contiene 47 exones y codifica la proteína llamada delangina, compuesta por 2.804 aminoácidos, perteneciente a la familia de las adherinas cromosómicas. La delangina parece facilitar las interacciones a larga distancia entre secuencias potenciadoras y promotoras.
La identificación de mutaciones en un solo alelo del gen NIPBL en individuos afectados es compatible con un patrón de herencia autosómico dominante. Hasta la fecha, todas las mutaciones detectadas dan lugar a proteínas truncadas con haploinsuficiencia, lo que explicaría la presencia de manifestaciones clínicas más graves en pacientes con grandes deleciones. Datos recientes apuntan a que el fenotipo SCdL leve es causado por mutaciones de cambio de sentido (missense).
Más recientemente, se han descubierto 2 nuevos genes relacionados con el SCdL: uno localizado en el cromosoma X, que se denominó SMC1A, y un tercero en el cromosoma 10, denominado SMC3, y del que hasta la fecha sólo se conoce a un paciente19.
AcondroplasiaClínicaLa acondroplasia (OMIM # 100800) es la forma hereditaria más común de las osteocondrodisplasias, con una incidencia de 1/15.000-1/40.000 nacidos vivos. Es un desorden autosómico dominante, con una penetrancia completa, y la mayoría de los casos presenta una mutación de novo. El fenotipo de la acondroplasia está relacionado con una formación anormal de los cartílagos, debido a mutaciones en el receptor del factor de crecimiento de fibroblastos 3 (FGFR3, del inglés fibroblast growth factor receptor 3). Los afectados presentan baja talla con tronco y extremidades cortas, macrocefalia, frente amplia y prominente, nariz corta con puente nasal deprimido y manos con dedos en forma de tridente. La inteligencia suele ser normal20,21.
El gen FGFR3 se identificó en 1994 por análisis de ligamiento en el cromosoma 4 (4p16.3). La proteína que codifica está implicada en diferentes mecanismos celulares (angiogenia, apoptosis, diferenciación, etc.)22.
Diagnóstico molecularEl 98% de los casos con acondroplasia presenta una mutación en este gen, en el dominio tirosincinasa. Es una mutación tipo "cambio de sentido" (missense), en la que una glicina se sustituye por una arginina en la posición 380 (Gly380Arg). Este porcentaje tan elevado de pacientes con la misma mutación permite realizar este estudio mediante enzimas de restricción, ya que el cambio de nucleótido producido altera el marco de lectura, y crea un nuevo punto de corte para una enzima de restricción (SfcI). El fragmento obtenido de ADN normal tiene un tamaño de 164 pb, mientras que el ADN mutado son 2 fragmentos de 109 y 55 pb, respectivamente (fig. 4). El diagnóstico también puede realizarse mediante secuenciación directa de ese fragmento de ADN, con la ventaja de que, además, podrían detectarse mutaciones en el 2% restante de los pacientes20.
 una a un alelo normal (164pb),y b) la otra a uno mutado, en el cual, por causa de la mutación, aparece un nuevo sitio de corte para la enzima SfcI, en el que aparecen 2 bandas, una de 109 pb y la otra de 55pb. En la línea 4, tenemos a un individuo normal, por lo tanto, sólo aparece la banda de 164pb. M: marcador estándar.")
Análisis de la restricción enzimática para el diagnóstico molecular de acondroplasia en gel de agarosa. En las líneas 1, 2 y 3 aparecen 3 bandas que corresponden: a) una a un alelo normal (164pb),y b) la otra a uno mutado, en el cual, por causa de la mutación, aparece un nuevo sitio de corte para la enzima SfcI, en el que aparecen 2 bandas, una de 109 pb y la otra de 55pb. En la línea 4, tenemos a un individuo normal, por lo tanto, sólo aparece la banda de 164pb. M: marcador estándar.


